
Mitochondrial DNA Haplogroups and Variants Predispose to Chagas Disease Cardiomyopathy

 , , , , , , , , , and add
Show full author list
, , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Collection
2.2. Heart Tissue and Blood DNA Isolation
2.3. Mitochondrial DNA Copy Number Quantification
2.4. Mitochondrial Haplogroup Determination
2.5. Variant Annotations
2.6. Statistical Analysis
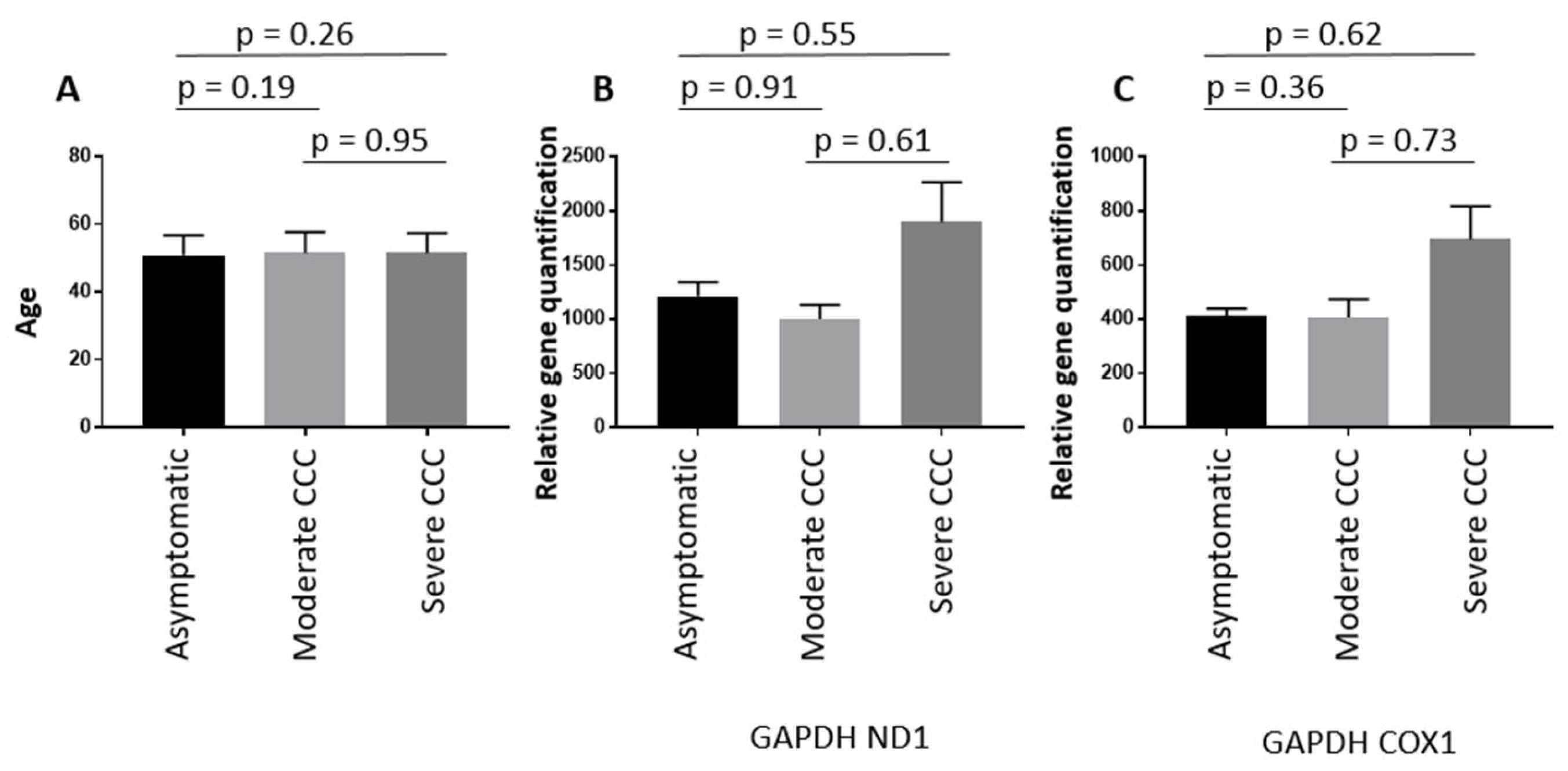
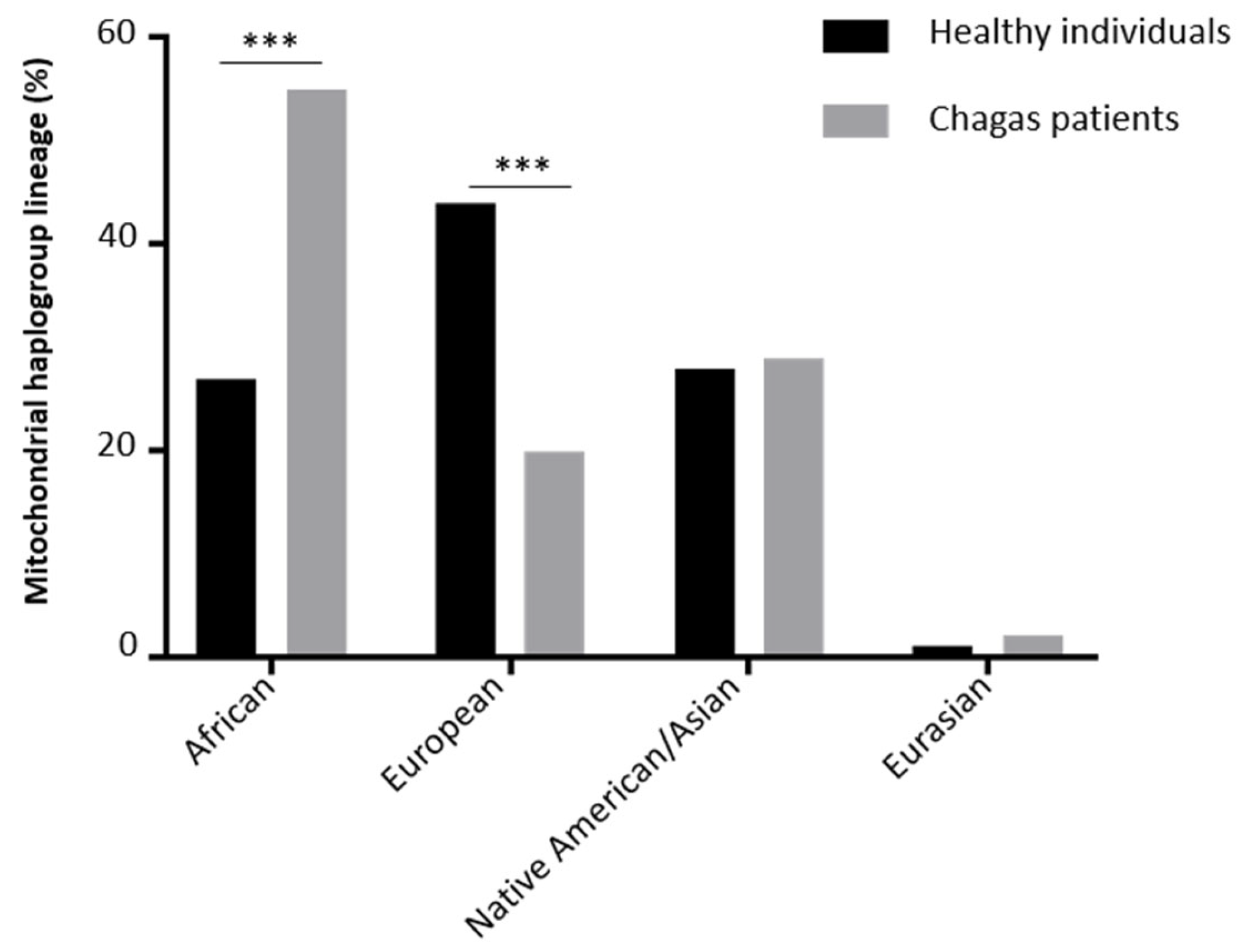
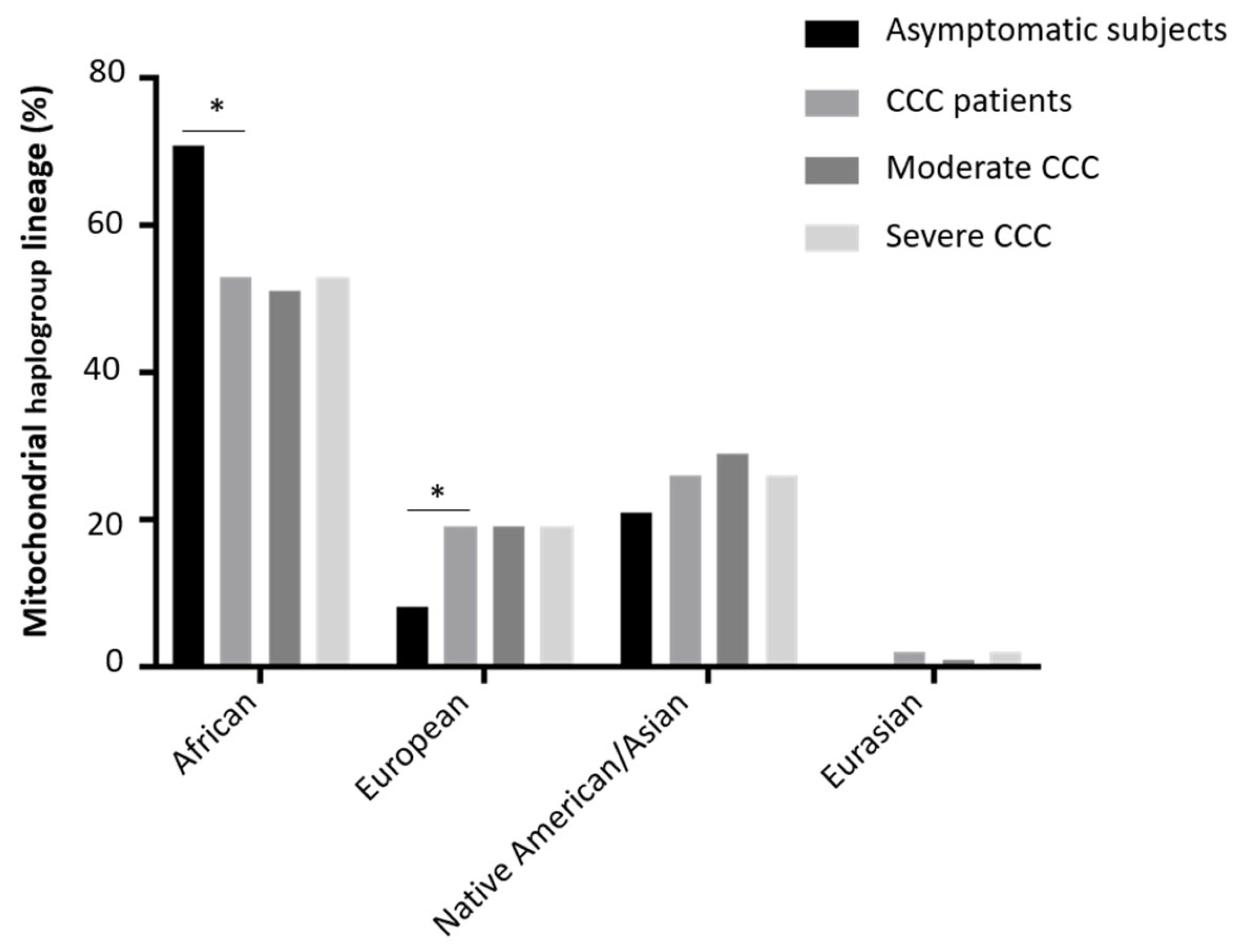
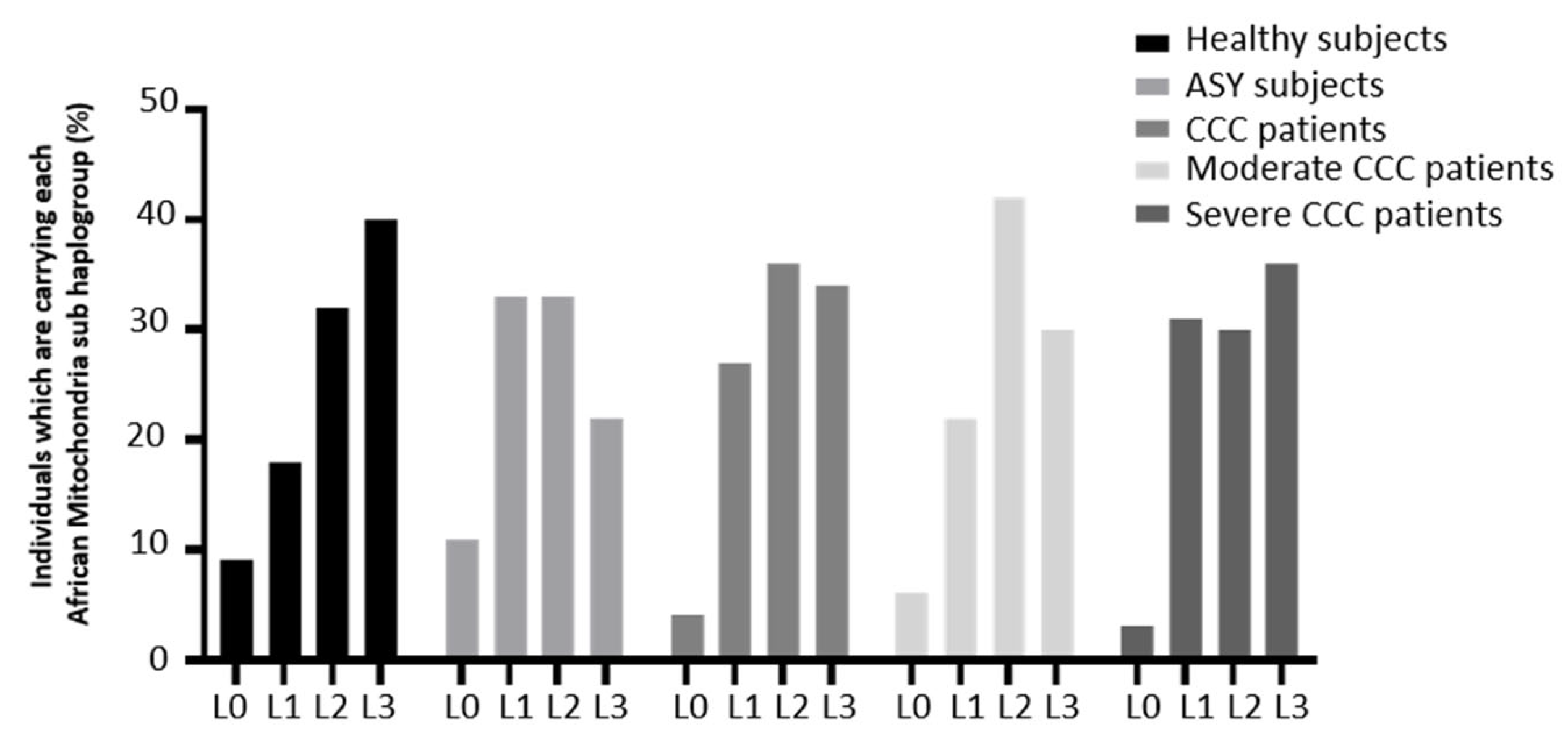
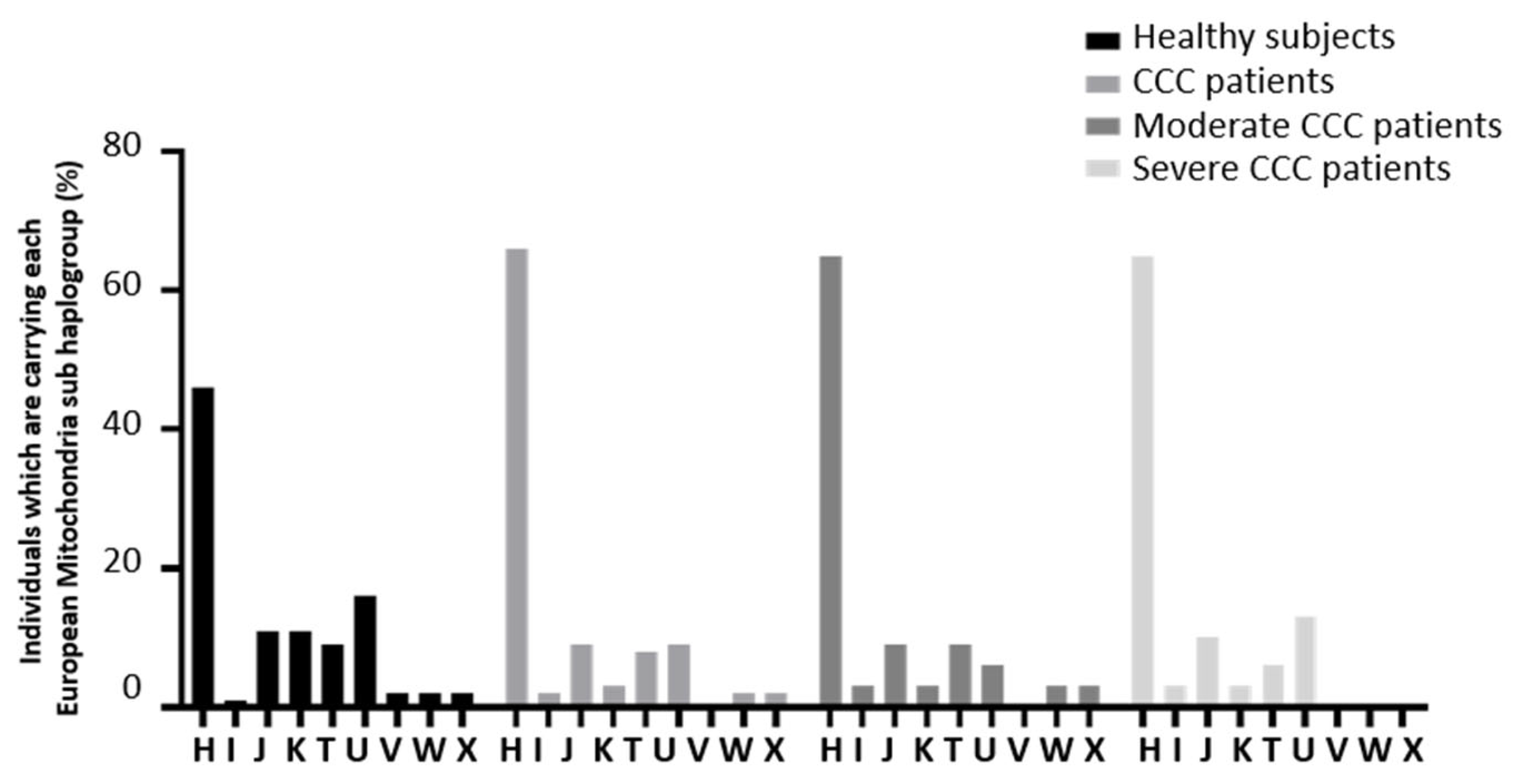
3. Results
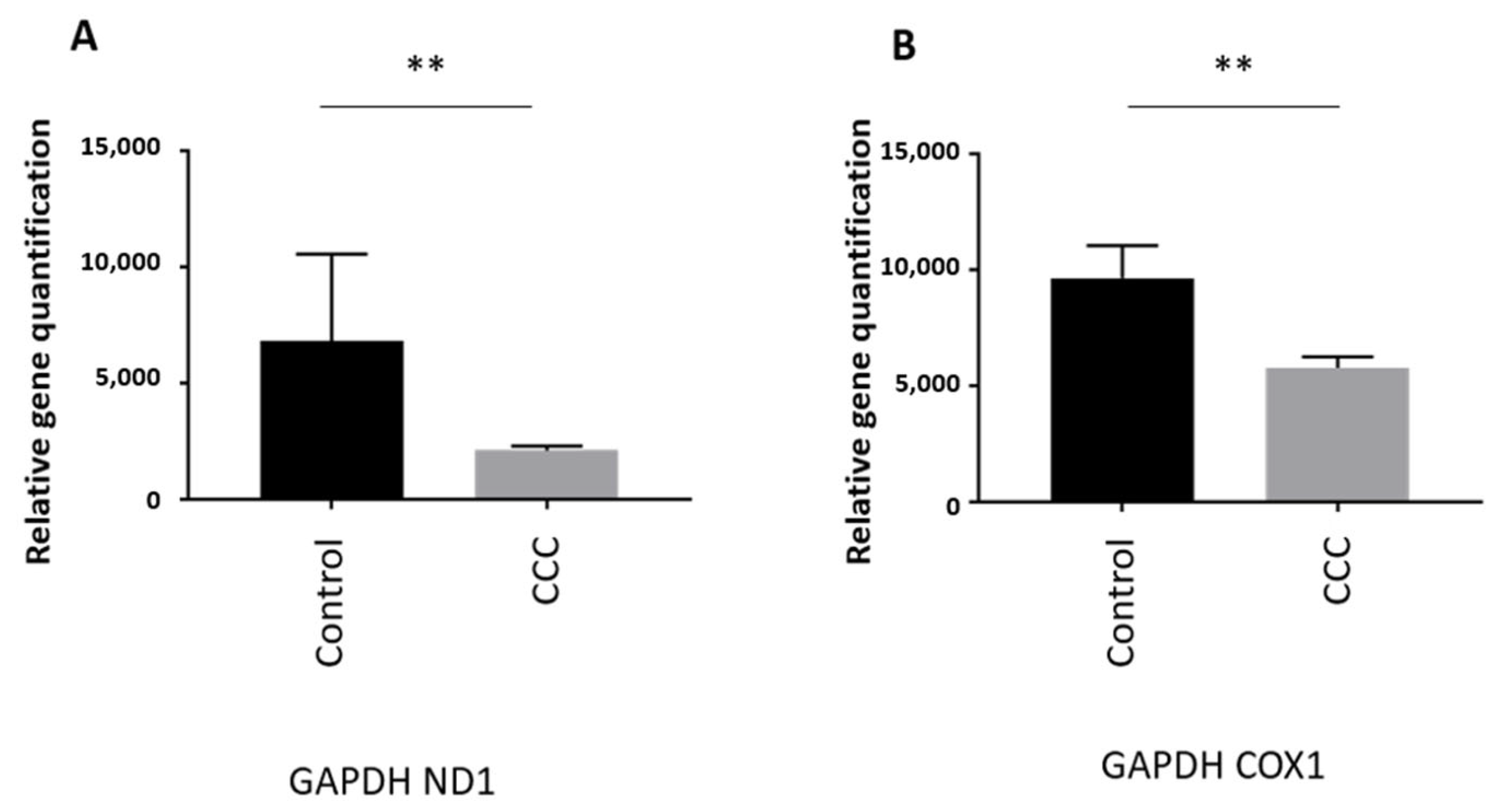
MtDNA Copy Number Is Reduced in Heart of Chagas Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134, e579–e646. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Brun, F.; Spezzacatene, A.; Sinagra, G.; Taylor, M.R. Genetic Causes of Dilated Cardiomyopathy. Prog. Pediatr. Cardiol. 2014, 37, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Bakalakos, A.; Ritsatos, K.; Anastasakis, A. Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: A Cardiomyopathy Clinic Doctor’s point of view. Hell. J. Cardiol. 2018, 59, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.M.; Lorenzini, M.; Cicerchia, M.; Ochoa, J.P.; Hey, T.M.; Sabater Molina, M.; Restrepo-Cordoba, M.A.; Dal Ferro, M.; Stolfo, D.; Johnson, R.; et al. Clinical Phenotypes and Prognosis of Dilated Cardiomyopathy Caused by Truncating Variants in the TTN Gene. Circ. Heart Fail. 2020, 13, e006832. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, S.E.; Cho, M.C. Clinical Implication of Genetic Testing in Dilated Cardiomyopathy. Int. J. Heart Fail. 2022, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.T.; Muchir, A.; Nagy, P.L.; Worman, H.J. LMNA cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis. Models Mech. 2011, 4, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Tabish, A.M.; Azzimato, V.; Alexiadis, A.; Buyandelger, B.; Knoll, R. Genetic epidemiology of titin-truncating variants in the etiology of dilated cardiomyopathy. Biophys. Rev. 2017, 9, 207–223. [Google Scholar] [CrossRef]
- Zak, R.; Rabinowitz, M.; Rajamanickam, C.; Merten, S.; Kwiatkowska-Patzer, B. Mitochondrial proliferation in cardiac hypertrophy. Basic. Res. Cardiol. 1980, 75, 171–178. [Google Scholar] [CrossRef]
- Goffart, S.; von Kleist-Retzow, J.C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, D.R.; Sugiana, C.; Salemi, R.; Kirby, D.M.; Worgan, L.; Ohtake, A.; Ryan, M.T. Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim. Biophys. Acta 2004, 1659, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679. [Google Scholar] [CrossRef]
- Larsen, S.; Diez-Sanchez, C.; Rabol, R.; Ara, I.; Dela, F.; Helge, J.W. Increased intrinsic mitochondrial function in humans with mitochondrial haplogroup H. Biochim. Biophys. Acta 2014, 1837, 226–231. [Google Scholar] [CrossRef]
- Perez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Morillo, C.A.; Marin-Neto, J.A.; Avezum, A.; Sosa-Estani, S.; Rassi, A., Jr.; Rosas, F.; Villena, E.; Quiroz, R.; Bonilla, R.; Britto, C.; et al. Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. N. Engl. J. Med. 2015, 373, 1295–1306. [Google Scholar] [CrossRef]
- Bestetti, R.B.; Muccillo, G. Clinical course of Chagas’ heart disease: A comparison with dilated cardiomyopathy. Int. J. Cardiol. 1997, 60, 187–193. [Google Scholar] [CrossRef]
- Mady, C.; Cardoso, R.H.; Barretto, A.C.; da Luz, P.L.; Bellotti, G.; Pileggi, F. Survival and predictors of survival in patients with congestive heart failure due to Chagas’ cardiomyopathy. Circulation 1994, 90, 3098–3102. [Google Scholar] [CrossRef]
- Higuchi, M.L.; De Morais, C.F.; Pereira Barreto, A.C.; Lopes, E.A.; Stolf, N.; Bellotti, G.; Pileggi, F. The role of active myocarditis in the development of heart failure in chronic Chagas’ disease: A study based on endomyocardial biopsies. Clin. Cardiol. 1987, 10, 665–670. [Google Scholar] [CrossRef]
- Pereira Barretto, A.C.; Mady, C.; Arteaga-Fernandez, E.; Stolf, N.; Lopes, E.A.; Higuchi, M.L.; Bellotti, G.; Pileggi, F. Right ventricular endomyocardial biopsy in chronic Chagas’ disease. Am. Heart J. 1986, 111, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Abel, L.C.; Rizzo, L.V.; Ianni, B.; Albuquerque, F.; Bacal, F.; Carrara, D.; Bocchi, E.A.; Teixeira, H.C.; Mady, C.; Kalil, J.; et al. Chronic Chagas’ disease cardiomyopathy patients display an increased IFN-gamma response to Trypanosoma cruzi infection. J. Autoimmun. 2001, 17, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, L.G.; Santos, R.H.; Ianni, B.M.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; Frade, A.; Donadi, E.; Dias, F.; Saba, B.; et al. Myocardial chemokine expression and intensity of myocarditis in Chagas cardiomyopathy are controlled by polymorphisms in CXCL9 and CXCL10. PLoS Negl. Trop. Dis. 2012, 6, e1867. [Google Scholar] [CrossRef] [PubMed]
- Talvani, A.; Rocha, M.O.; Barcelos, L.S.; Gomes, Y.M.; Ribeiro, A.L.; Teixeira, M.M. Elevated concentrations of CCL2 and tumor necrosis factor-alpha in chagasic cardiomyopathy. Clin. Infect. Dis. 2004, 38, 943–950. [Google Scholar] [CrossRef]
- Teixeira, M.M.; Gazzinelli, R.T.; Silva, J.S. Chemokines, inflammation and Trypanosoma cruzi infection. Trends Parasitol. 2002, 18, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Laugier, L.; Ferreira, L.R.P.; Ferreira, F.M.; Cabantous, S.; Frade, A.F.; Nunes, J.P.; Ribeiro, R.A.; Brochet, P.; Teixeira, P.C.; Santos, R.H.B.; et al. miRNAs may play a major role in the control of gene expression in key pathobiological processes in Chagas disease cardiomyopathy. PLoS Negl. Trop. Dis. 2020, 14, e0008889. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Neto, E.; Teixeira, P.C.; Fonseca, S.G.; Bilate, A.M.; Kalil, J. Myocardial gene and protein expression profiles after autoimmune injury in Chagas’ disease cardiomyopathy. Autoimmun. Rev. 2011, 10, 163–165. [Google Scholar] [CrossRef]
- Teixeira, P.C.; Ducret, A.; Langen, H.; Nogoceke, E.; Santos, R.H.B.; Silva Nunes, J.P.; Benvenuti, L.; Levy, D.; Bydlowski, S.P.; Bocchi, E.A.; et al. Impairment of Multiple Mitochondrial Energy Metabolism Pathways in the Heart of Chagas Disease Cardiomyopathy Patients. Front. Immunol. 2021, 12, 755782. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.C.; Santos, R.H.; Fiorelli, A.I.; Bilate, A.M.; Benvenuti, L.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Selective decrease of components of the creatine kinase system and ATP synthase complex in chronic Chagas disease cardiomyopathy. PLoS Negl. Trop. Dis. 2011, 5, e1205. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.P.S.; Andrieux, P.; Brochet, P.; Almeida, R.R.; Kitano, E.; Honda, A.K.; Iwai, L.K.; Andrade-Silva, D.; Goudenege, D.; Alcantara Silva, K.D.; et al. Co-Exposure of Cardiomyocytes to IFN-gamma and TNF-alpha Induces Mitochondrial Dysfunction and Nitro-Oxidative Stress: Implications for the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Front. Immunol. 2021, 12, 755862. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C., Jr.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Zicker, F.; Smith, P.G.; Netto, J.C.; Oliveira, R.M.; Zicker, E.M. Physical activity, opportunity for reinfection, and sibling history of heart disease as risk factors for Chagas’ cardiopathy. Am. J. Trop. Med. Hyg. 1990, 43, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Ouarhache, M.; Marquet, S.; Frade, A.F.; Ferreira, A.M.; Ianni, B.; Almeida, R.R.; Nunes, J.P.S.; Ferreira, L.R.P.; Rigaud, V.O.; Candido, D.; et al. Rare Pathogenic Variants in Mitochondrial and Inflammation-Associated Genes May Lead to Inflammatory Cardiomyopathy in Chagas Disease. J. Clin. Immunol. 2021, 41, 1048–1063. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef]
- Naslavsky, M.S.; Scliar, M.O.; Yamamoto, G.L.; Wang, J.Y.T.; Zverinova, S.; Karp, T.; Nunes, K.; Ceroni, J.R.M.; de Carvalho, D.L.; da Silva Simoes, C.E.; et al. Whole-genome sequencing of 1171 elderly admixed individuals from Sao Paulo, Brazil. Nat. Commun. 2022, 13, 1004. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Herzeel, C.; Costanza, P.; Decap, D.; Fostier, J.; Verachtert, W. elPrep 4: A multithreaded framework for sequence analysis. PLoS ONE 2019, 14, e0209523. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Goudenege, D.; Bris, C.; Hoffmann, V.; Desquiret-Dumas, V.; Jardel, C.; Rucheton, B.; Bannwarth, S.; Paquis-Flucklinger, V.; Lebre, A.S.; Colin, E.; et al. eKLIPse: A sensitive tool for the detection and quantification of mitochondrial DNA deletions from next-generation sequencing data. Genet. Med. 2019, 21, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Castellana, S.; Ronai, J.; Mazza, T. MitImpact: An exhaustive collection of pre-computed pathogenicity predictions of human mitochondrial non-synonymous variants. Hum. Mutat. 2015, 36, E2413–E2422. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.C.; Iwai, L.K.; Kuramoto, A.C.; Honorato, R.; Fiorelli, A.; Stolf, N.; Kalil, J.; Cunha-Neto, E. Proteomic inventory of myocardial proteins from patients with chronic Chagas’ cardiomyopathy. Braz. J. Med. Biol. Res. 2006, 39, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Picard, M. Blood mitochondrial DNA copy number: What are we counting? Mitochondrion 2021, 60, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, K.; Sundquist, J.; Palmer, K.; Memon, A.A. Role of mitochondrial DNA copy number in incident cardiovascular diseases and the association between cardiovascular disease and type 2 diabetes: A follow-up study on middle-aged women. Atherosclerosis 2022, 341, 58–62. [Google Scholar] [CrossRef]
- Nah, J. The Role of Alternative Mitophagy in Heart Disease. Int. J. Mol. Sci. 2023, 24, 6362. [Google Scholar] [CrossRef]
- Dutta, D.; Calvani, R.; Bernabei, R.; Leeuwenburgh, C.; Marzetti, E. Contribution of impaired mitochondrial autophagy to cardiac aging: Mechanisms and therapeutic opportunities. Circ. Res. 2012, 110, 1125–1138. [Google Scholar] [CrossRef]
- Romano, P.S.; Cueto, J.A.; Casassa, A.F.; Vanrell, M.C.; Gottlieb, R.A.; Colombo, M.I. Molecular and cellular mechanisms involved in the Trypanosoma cruzi/host cell interplay. IUBMB Life 2012, 64, 387–396. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Brochet, P.; Ianni, B.M.; Laugier, L.; Frade, A.F.; Silva Nunes, J.P.; Teixeira, P.C.; Mady, C.; Ferreira, L.R.P.; Ferre, Q.; Santos, R.H.B.; et al. Epigenetic regulation of transcription factor binding motifs promotes Th1 response in Chagas disease cardiomyopathy. Front. Immunol. 2022, 13, 958200. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.M.; Aidt, F.H.; Hedley, P.L.; Jensen, M.K.; Havndrup, O.; Kanters, J.K.; Moolman-Smook, J.C.; Larsen, S.O.; Bundgaard, H.; Christiansen, M. Mitochondrial haplogroups modify the risk of developing hypertrophic cardiomyopathy in a Danish population. PLoS ONE 2013, 8, e71904. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, P.; Rani, B.; Sundaravadivel, P.; Vanniarajan, A.; Indumathi, K.P.; Khan, N.A.; Dhandapany, P.S.; Rani, D.S.; Tamang, R.; Bahl, A.; et al. Mitochondrial genome variations in idiopathic dilated cardiomyopathy. Mitochondrion 2019, 48, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Caggiano, M.; Barallobre-Barreiro, J.; Rego-Perez, I.; Crespo-Leiro, M.G.; Paniagua, M.J.; Grille, Z.; Blanco, F.J.; Domenech, N. Mitochondrial DNA haplogroup H as a risk factor for idiopathic dilated cardiomyopathy in Spanish population. Mitochondrion 2013, 13, 263–268. [Google Scholar] [CrossRef]
- McManus, M.J.; Picard, M.; Chen, H.W.; De Haas, H.J.; Potluri, P.; Leipzig, J.; Towheed, A.; Angelin, A.; Sengupta, P.; Morrow, R.M.; et al. Mitochondrial DNA Variation Dictates Expressivity and Progression of Nuclear DNA Mutations Causing Cardiomyopathy. Cell Metab. 2019, 29, 78–90.e5. [Google Scholar] [CrossRef]
- Garret, P.; Bris, C.; Procaccio, V.; Amati-Bonneau, P.; Vabres, P.; Houcinat, N.; Tisserant, E.; Feillet, F.; Bruel, A.L.; Quere, V.; et al. Deciphering exome sequencing data: Bringing mitochondrial DNA variants to light. Hum. Mutat. 2019, 40, 2430–2443. [Google Scholar] [CrossRef]
- Yahata, N.; Matsumoto, Y.; Omi, M.; Yamamoto, N.; Hata, R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m.13513G>A mutation. Sci. Rep. 2017, 7, 15557. [Google Scholar] [CrossRef]
- Gammage, P.A.; Minczuk, M. Enhanced Manipulation of Human Mitochondrial DNA Heteroplasmy In Vitro Using Tunable mtZFN Technology. Methods Mol. Biol. 2018, 1867, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef]
- Hussain, S.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef] [PubMed]
- Bian, W.P.; Chen, Y.L.; Luo, J.J.; Wang, C.; Xie, S.L.; Pei, D.S. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth. Biol. 2019, 8, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef] [PubMed]
- Kar, B.; Castillo, S.R.; Sabharwal, A.; Clark, K.J.; Ekker, S.C. Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities. Int. J. Mol. Sci. 2023, 24, 5798. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.J.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol. Genet. Metab. 2012, 105, 607–614. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- Rotig, A.; Appelkvist, E.L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; Lebideau, M.; Dallner, G.; Munnich, A.; Ernster, L.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–395. [Google Scholar] [CrossRef]
- Di Giovanni, S.; Mirabella, M.; Spinazzola, A.; Crociani, P.; Silvestri, G.; Broccolini, A.; Tonali, P.; Di Mauro, S.; Servidei, S. Coenzyme Q10 reverses pathological phenotype and reduces apoptosis in familial CoQ10 deficiency. Neurology 2001, 57, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Saneto, R.; Falk, M.J.; Anselm, I.; Cohen, B.H.; Haas, R.; Medicine Society, T.M. A modern approach to the treatment of mitochondrial disease. Curr. Treat. Options Neurol. 2009, 11, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, 2012, CD004426. [Google Scholar] [CrossRef]
- Almannai, M.; El-Hattab, A.W. Nitric Oxide Deficiency in Mitochondrial Disorders: The Utility of Arginine and Citrulline. Front. Mol. Neurosci. 2021, 14, 682780. [Google Scholar] [CrossRef] [PubMed]
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 11338. [Google Scholar] [CrossRef] [PubMed]
- Laugier, L.; Frade, A.F.; Ferreira, F.M.; Baron, M.A.; Teixeira, P.C.; Cabantous, S.; Ferreira, L.R.P.; Louis, L.; Rigaud, V.O.C.; Gaiotto, F.A.; et al. Whole-Genome Cardiac DNA Methylation Fingerprint and Gene Expression Analysis Provide New Insights in the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Clin. Infect. Dis. 2017, 65, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Crisosto, C.; Pennanen, C.; Vasquez-Trincado, C.; Morales, P.E.; Bravo-Sagua, R.; Quest, A.F.G.; Chiong, M.; Lavandero, S. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017, 14, 342–360. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, D.; Dong, X.; Zhang, X.; Liu, J.; Peng, L.; Meng, B.; Hua, Q.; Pei, X.; Zhao, L.; et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. Sci. China Life Sci. 2022, 65, 1198–1212. [Google Scholar] [CrossRef]
- Chen, W.J.; Cheng, Y.; Li, W.; Dong, X.K.; Wei, J.L.; Yang, C.H.; Jiang, Y.H. Quercetin Attenuates Cardiac Hypertrophy by Inhibiting Mitochondrial Dysfunction Through SIRT3/PARP-1 Pathway. Front. Pharmacol. 2021, 12, 739615. [Google Scholar] [CrossRef]
- Chen, J.; Chen, S.; Zhang, B.; Liu, J. SIRT3 as a potential therapeutic target for heart failure. Pharmacol. Res. 2021, 165, 105432. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Haplogroup Lineage | End stage Heart Tissue Samples (CCC) | Healthy Hearts of Organ Donors (Control) |
|---|---|---|
| African | 19 (73%) | 4 (67%) |
| European | 4 (15%) | 0 (0%) |
| Native American/Asian | 2 (8%) | 1 (17%) |
| Eurasian | 1 (4%) | 1 (17%) |
| Groups | p Value | Relative Risk |
|---|---|---|
| Healthy individuals versus Chagas patients | 0.01 | 0.76 |
| Healthy individuals versus moderate Chagas cardiomyopathy patients | 0.06 | 0.85 |
| Healthy individuals versus severe Chagas cardiomyopathy patients | 0.07 | 0.86 |
| Pos | Ref | Alt | Csq | Gene Symbol | dbSNP ID | Uniprot Name | Uniprot ID | AA Pos | AA Ref | AA Alt | Described as Pathogenic in the following Databases. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3565 | A | C | RNA | MT-ND1 | NU1M | P03886 | 87 | T | P | PolyPhen2, SIFT, PROVEAN, CADD, PhDSNP, SP, COVEC_WMV, MtoolBox | |
| 3579 | A | C | RNA | MT-ND1 | NU1M | P03886 | 91 | M | I | CADD, PhDSNP, MutationTaster, SNPDryad, Condel | |
| 5262 | G | A | RNA | MT-ND2 | NU2M | P03891 | 265 | A | T | CADD, PANTHER, Condel, | |
| 6571 | C | T | RNA | MT-CO1 | COX1 | P00395 | 223 | A | V | PROVEAN, EFIN_SP, CADD, PhDSNP, MutationTaster, Condel, PolyPhen2 transf | |
| 6873 | C | T | RNA | MT-CO1 | COX1 | P00395 | 324 | L | F | PolyPhen2, SIFT, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, MutationTaster, COVEC_WMV, MtoolBox, Mutatiossessor transf | |
| 6899 | G | C | RNA | MT-CO1 | rs1556423194 | COX1 | P00395 | 332 | M | I | Condel, PolyPhen2 transf, SIFT transf |
| 6909 | G | T | RNA | MT-CO1 | COX1 | P00395 | 336 | A | S | SIFT, CADD, PhDSNP | |
| 6915 | G | A | RNA | MT-CO1 | rs1603220687 | COX1 | P00395 | 338 | V | M | Condel, PolyPhen2 transf, SIFT transf |
| 7008 | G | T | RNA | MT-CO1 | COX1 | P00395 | 369 | D | Y | PolyPhen2, SIFT, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 7072 | T | C | RNA | MT-CO1 | rs1603220760 | COX1 | P00395 | 390 | M | T | PolyPhen2, SIFT, EFIN_HD, CADD, PhDSNP, SP, MutationTaster, COVEC_WMV, MtoolBox |
| 7868 | C | T | RNA | MT-CO2 | rs1556423357 | COX2 | P00403 | 95 | L | F | PROVEAN, CADD, PANTHER, PhDSNP, Condel, PolyPhen2 transf |
| 8021 | A | G | RNA | MT-CO2 | rs1603221261 | COX2 | P00403 | 146 | I | V | Condel, PolyPhen2 transf |
| 8401 | A | C | INT | MT-ATP8 | ATP8 | P03928 | 12 | M | I | Condel, SIFT transf | |
| 8405 | A | G | INT | MT-ATP8 | rs1603221462 | ATP8 | P03928 | 14 | T | A | Condel, PolyPhen2 transf |
| 8527 | A | G | RNA | MT-ATP6 | rs878853003 | ATP6 | P00846 | 1 | M | V | PolyPhen2, MtoolBox |
| 8558 | C | G | RNA | MT-ATP8 | ATP8 | P03928 | 65 | P | A | PolyPhen2, FatHmm, PROVEAN, CADD, SP, COVEC_WMV, MtoolBox | |
| 8558 | C | G | RNA | MT-ATP8 | ATP6 | P00846 | 11 | A | G | CADD, PANTHER, PhDSNP | |
| 8566 | A | G | RNA | MT-ATP6 | rs3020563 | ATP6 | P00846 | 14 | I | V | SP, Condel |
| 8568 | C | T | RNA | MT-ATP8 | rs1603221589 | ATP8 | P03928 | 68 | S | F | PolyPhen2, FatHmm, EFIN_HD, CADD, SP, MtoolBox |
| 8618 | T | C | RNA | MT-ATP6 | rs28358885 | ATP6 | P00846 | 31 | I | T | Condel |
| 8632 | T | C | RNA | MT-ATP6 | rs1603221654 | ATP6 | P00846 | 36 | Y | H | PANTHER, Condel, PolyPhen2 transf |
| 8677 | A | C | RNA | MT-ATP6 | ATP6 | P00846 | 51 | K | Q | Condel, PolyPhen2 transf | |
| 8714 | C | T | RNA | MT-ATP6 | rs1603221724 | ATP6 | P00846 | 63 | T | I | FatHmm, PANTHER, Condel |
| 8795 | A | C | RNA | MT-ATP6 | ATP6 | P00846 | 90 | H | P | PolyPhen2, SIFT, PROVEAN, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox | |
| 8854 | G | A | RNA | MT-ATP6 | rs386829055 | ATP6 | P00846 | 110 | A | T | SIFT, CADD, PANTHER, PhDSNP, Condel |
| 8860 | A | G | RNA | MT-ATP6 | rs2001031 | ATP6 | P00846 | 112 | T | A | PROVEAN, PhDSNP, SP, Condel, PolyPhen2 transf |
| 8870 | T | C | RNA | MT-ATP6 | rs1556423560 | ATP6 | P00846 | 115 | M | T | Condel, PolyPhen2 transf |
| 8875 | T | C | RNA | MT-ATP6 | rs201123510 | ATP6 | P00846 | 117 | F | L | Condel, PolyPhen2 transf |
| 9261 | A | C | RNA | MT-CO3 | COX3 | P00414 | 19 | T | P | PolyPhen2, PROVEAN, EFIN_SP, CADD, PhDSNP, SP, SNPDryad, MtoolBox | |
| 9325 | T | C | RNA | MT-CO3 | rs879000531 | COX3 | P00414 | 40 | M | T | Condel, PolyPhen2 transf |
| 9717 | C | G | RNA | MT-CO3 | COX3 | P00414 | 171 | L | V | Condel, PolyPhen2 transf | |
| 9861 | T | C | RNA | MT-CO3 | rs878853060 | COX3 | P00414 | 219 | F | L | Condel, PolyPhen2 transf, SIFT transf |
| 9877 | T | A | RNA | MT-CO3 | COX3 | P00414 | 224 | M | K | MutationTaster, Condel | |
| 9880 | T | A | RNA | MT-CO3 | COX3 | P00414 | 225 | F | Y | Condel, PolyPhen2 transf | |
| 9896 | A | C | RNA | MT-CO3 | COX3 | P00414 | 230 | K | N | Condel, PolyPhen2 transf | |
| 9924 | T | G | RNA | MT-CO3 | COX3 | P00414 | 240 | W | G | PolyPhen2, PROVEAN, Mutatiossessor, EFIN_HD, CADD, VEST, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox | |
| 10677 | G | A | RNA | MT-ND4L | rs1603222944 | NU4LM | P03901 | 70 | E | K | FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, Condel, MtoolBox, Mutatiossessor transf |
| 10750 | A | G | RNA | MT-ND4L | rs372297272 | NU4LM | P03901 | 94 | N | S | PROVEAN, PhDSNP, MutationTaster, Condel |
| 10775 | G | A | RNA | MT-ND4 | rs879015842 | NU4M | P03905 | 6 | V | I | Condel, PolyPhen2 transf, SIFT transf |
| 10808 | C | T | RNA | MT-ND4 | rs2068723560 | NU4M | P03905 | 17 | L | F | CADD, Condel, MtoolBox |
| 10866 | T | C | RNA | MT-ND4 | rs1603222994 | NU4M | P03905 | 36 | I | T | Condel |
| 10972 | A | C | RNA | MT-ND4 | NU4M | P03905 | 71 | W | C | PolyPhen2, SIFT, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 11129 | A | G | RNA | MT-ND4 | rs1603223122 | NU4M | P03905 | 124 | T | A | Condel, SIFT transf |
| 11963 | G | A | RNA | MT-ND4 | rs201803948 | NU4M | P03905 | 402 | V | I | Condel, PolyPhen2 transf, SIFT transf |
| 12341 | C | A | RNA | MT-ND5 | NU5M | P03915 | 2 | T | N | Condel, PolyPhen2 transf | |
| 12346 | C | T | RNA | MT-ND5 | NU5M | P03915 | 4 | H | Y | Condel, PolyPhen2 transf, SIFT transf | |
| 12368 | C | T | RNA | MT-ND5 | NU5M | P03915 | 11 | T | I | FatHmm, PROVEAN, Condel, PolyPhen2 transf | |
| 12403 | C | T | RNA | MT-ND5 | NU5M | P03915 | 23 | L | F | Condel, PolyPhen2 transf | |
| 12802 | A | G | RNA | MT-ND5 | NU5M | P03915 | 156 | S | G | Condel | |
| 13034 | T | C | RNA | MT-ND5 | NU5M | P03915 | 233 | L | P | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13118 | T | A | RNA | MT-ND5 | NU5M | P03915 | 261 | I | N | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13145 | G | A | RNA | MT-ND5 | NU5M | P03915 | 270 | S | N | Condel | |
| 13226 | A | C | RNA | MT-ND5 | NU5M | P03915 | 297 | D | A | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, MutationTaster, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13303 | C | T | RNA | MT-ND5 | NU5M | P03915 | 323 | H | Y | PANTHER, PhDSNP, Condel, SIFT transf | |
| 13466 | G | A | RNA | MT-ND5 | NU5M | P03915 | 377 | S | N | FatHmm, PANTHER, Condel | |
| 13476 | A | T | RNA | MT-ND5 | NU5M | P03915 | 380 | L | F | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, EFIN_HD, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, Mutatiossessor transf | |
| 13517 | A | T | RNA | MT-ND5 | NU5M | P03915 | 394 | H | L | PANTHER, Condel, PolyPhen2 transf | |
| 13600 | T | A | RNA | MT-ND5 | NU5M | P03915 | 422 | Y | N | PolyPhen2, FatHmmW, FatHmm, PROVEAN, Mutatiossessor, EFIN_SP, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, APOGEE, DEOGEN2, Mutatiossessor transf | |
| 13614 | A | T | RNA | MT-ND5 | NU5M | P03915 | 426 | M | I | Condel | |
| 13618 | C | T | RNA | MT-ND5 | NU5M | P03915 | 428 | L | F | Condel, MtoolBox | |
| 13651 | A | G | RNA | MT-ND5 | rs1569484594 | NU5M | P03915 | 439 | T | A | CADD, Condel, MtoolBox |
| 13763 | C | A | RNA | MT-ND5 | NU5M | P03915 | 476 | S | Y | PolyPhen2, PROVEAN, CADD, PANTHER, SP, Condel, MtoolBox, SIFT transf | |
| 13804 | G | A | RNA | MT-ND5 | rs1603224360 | NU5M | P03915 | 490 | A | T | PolyPhen2, PROVEAN, CADD, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox |
| 13816 | A | C | RNA | MT-ND5 | NU5M | P03915 | 494 | T | P | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, CADD, PANTHER, PhDSNP, SP, SNPDryad, COVEC_WMV, MtoolBox, Mutatiossessor transf | |
| 13886 | T | C | RNA | MT-ND5 | rs28359182 | NU5M | P03915 | 517 | L | P | PhDSNP, Condel |
| 14170 | A | T | RNA | MT-ND6 | NU6M | P03923 | 168 | I | M | PolyPhen2, MtoolBox | |
| 14172 | T | G | RNA | MT-ND6 | NU6M | P03923 | 168 | I | L | PolyPhen2, CADD, MtoolBox | |
| 14280 | A | G | RNA | MT-ND6 | NU6M | P03923 | 132 | S | P | FatHmm, PhDSNP, Condel | |
| 14562 | C | T | RNA | MT-ND6 | NU6M | P03923 | 38 | V | I | Condel | |
| 14757 | T | C | RNA | MT-CYB | rs1603224859 | CYB | P00156 | 4 | M | T | Condel, PolyPhen2 transf |
| 14793 | A | G | RNA | MT-CYB | rs2853504 | CYB | P00156 | 16 | H | R | PhDSNP, SP, Condel, PolyPhen2 transf |
| 14798 | T | C | RNA | MT-CYB | rs28357681 | CYB | P00156 | 18 | F | L | Condel, PolyPhen2 transf |
| 14871 | T | C | RNA | MT-CYB | rs28660155 | CYB | P00156 | 42 | I | T | PROVEAN, PhDSNP, Condel, PolyPhen2 transf |
| 14873 | C | A | RNA | MT-CYB | CYB | P00156 | 43 | L | I | Condel | |
| 14881 | C | G | RNA | MT-CYB | CYB | P00156 | 45 | I | M | PolyPhen2, FatHmm, PANTHER, PhDSNP, SP, MtoolBox | |
| 15077 | G | A | RNA | MT-CYB | rs201943501 | CYB | P00156 | 111 | E | K | PolyPhen2, PROVEAN, CADD, PhDSNP, SNPDryad, MtoolBox |
| 15596 | G | A | RNA | MT-CYB | rs1603225369 | CYB | P00156 | 284 | V | I | Condel |
| 15617 | G | A | RNA | MT-CYB | rs1556424625 | CYB | P00156 | 291 | V | I | PolyPhen2, CADD, PhDSNP, SP, COVEC_WMV, MtoolBox |
| 15664 | C | A | RNA | MT-CYB | rs1603225414 | CYB | P00156 | 306 | I | M | CADD, Condel |
| 15699 | G | A | RNA | MT-CYB | CYB | P00156 | 318 | R | H | PolyPhen2, FatHmm, PROVEAN, Mutatiossessor, EFIN_HD, CADD, PANTHER, PhDSNP, SNPDryad, COVEC_WMV, MtoolBox, Mutatiossessor transf | |
| 15725 | C | T | RNA | MT-CYB | rs1603225438 | CYB | P00156 | 327 | L | F | PANTHER, Condel |
| 15734 | G | A | RNA | MT-CYB | rs386829259 | CYB | P00156 | 330 | A | T | PhDSNP, Condel |
| 15777 | G | A | RNA | MT-CYB | rs879182710 | CYB | P00156 | 344 | S | N | CADD, PhDSNP, Condel |
| 15812 | G | A | RNA | MT-CYB | rs200336777 | CYB | P00156 | 356 | V | M | EFIN_SP, MutationTaster, Condel |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallardo, F.; Brochet, P.; Goudenège, D.; Nunes, J.P.S.; Andrieux, P.; Ianni, B.M.; Frade, A.F.; Mady, C.; Santos, R.H.B.; Kuramoto, A.; et al. Mitochondrial DNA Haplogroups and Variants Predispose to Chagas Disease Cardiomyopathy. Hearts 2023, 4, 97-117. https://doi.org/10.3390/hearts4040013
Gallardo F, Brochet P, Goudenège D, Nunes JPS, Andrieux P, Ianni BM, Frade AF, Mady C, Santos RHB, Kuramoto A, et al. Mitochondrial DNA Haplogroups and Variants Predispose to Chagas Disease Cardiomyopathy. Hearts. 2023; 4(4):97-117. https://doi.org/10.3390/hearts4040013
Chicago/Turabian StyleGallardo, Frédéric, Pauline Brochet, David Goudenège, João Paulo Silva Nunes, Pauline Andrieux, Barbara Maria Ianni, Amanda Farage Frade, Charles Mady, Ronaldo Honorato Barros Santos, Andreia Kuramoto, and et al. 2023. "Mitochondrial DNA Haplogroups and Variants Predispose to Chagas Disease Cardiomyopathy" Hearts 4, no. 4: 97-117. https://doi.org/10.3390/hearts4040013