Synthesis and Characterization of B4C-Based Multifunctional Nanoparticles for Boron Neutron Capture Therapy Applications

 , , ,
, , ,  , , ,
, , ,  , and
, and 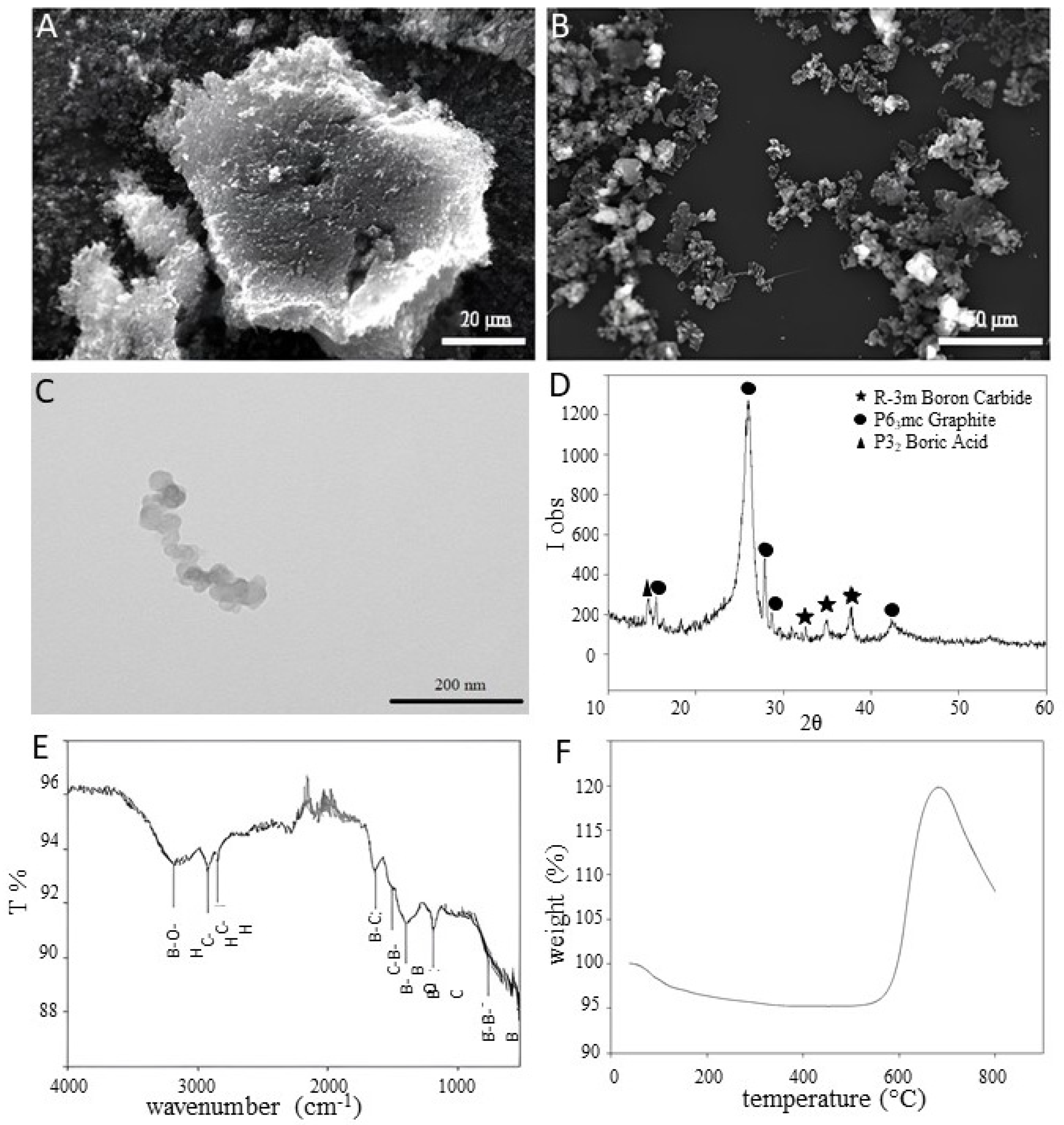{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of FeBNPs
2.2. Nanoparticles’ Characterization
2.3. Characterization of NPs–HeLa Cells Interaction
3. Results
3.1. B4C NPs’ Characterization
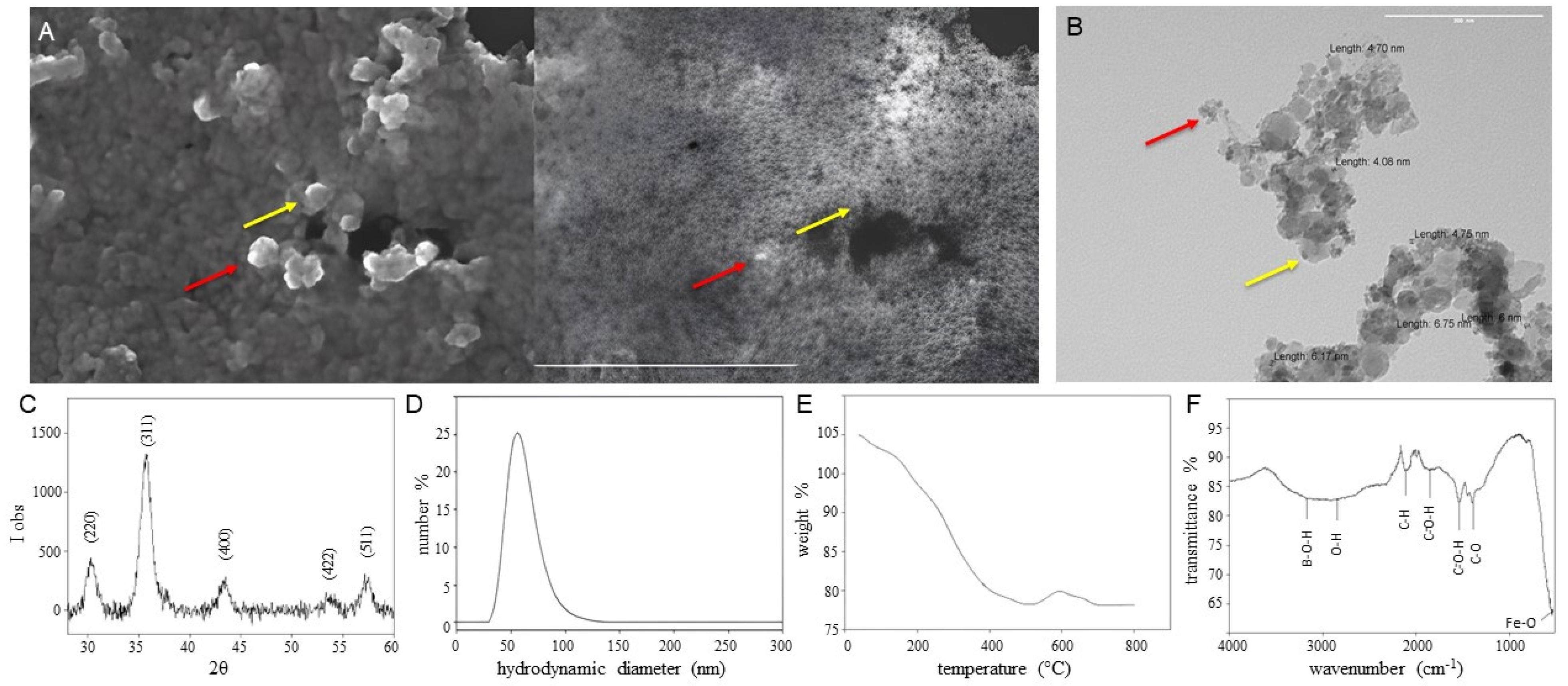
3.2. FeBNPs’ Characterization
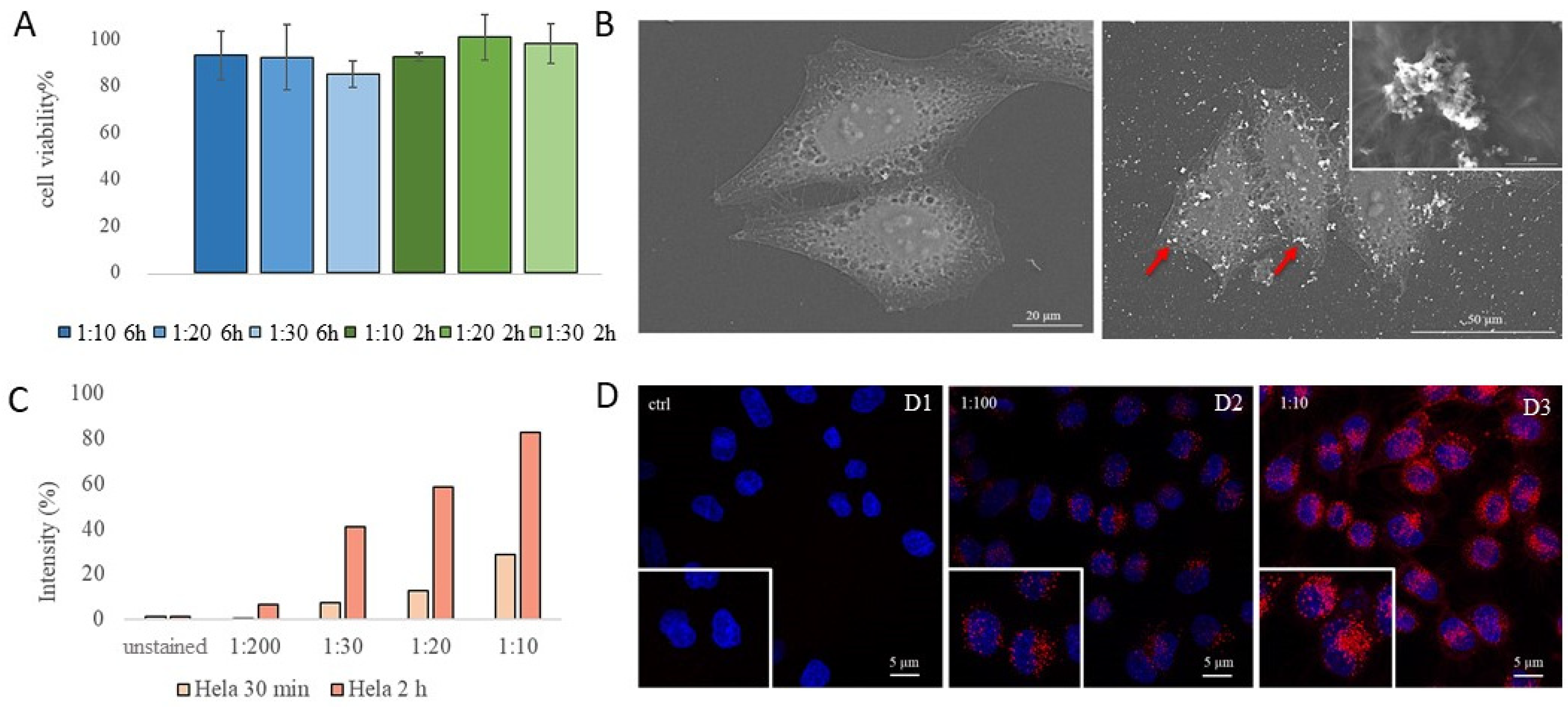
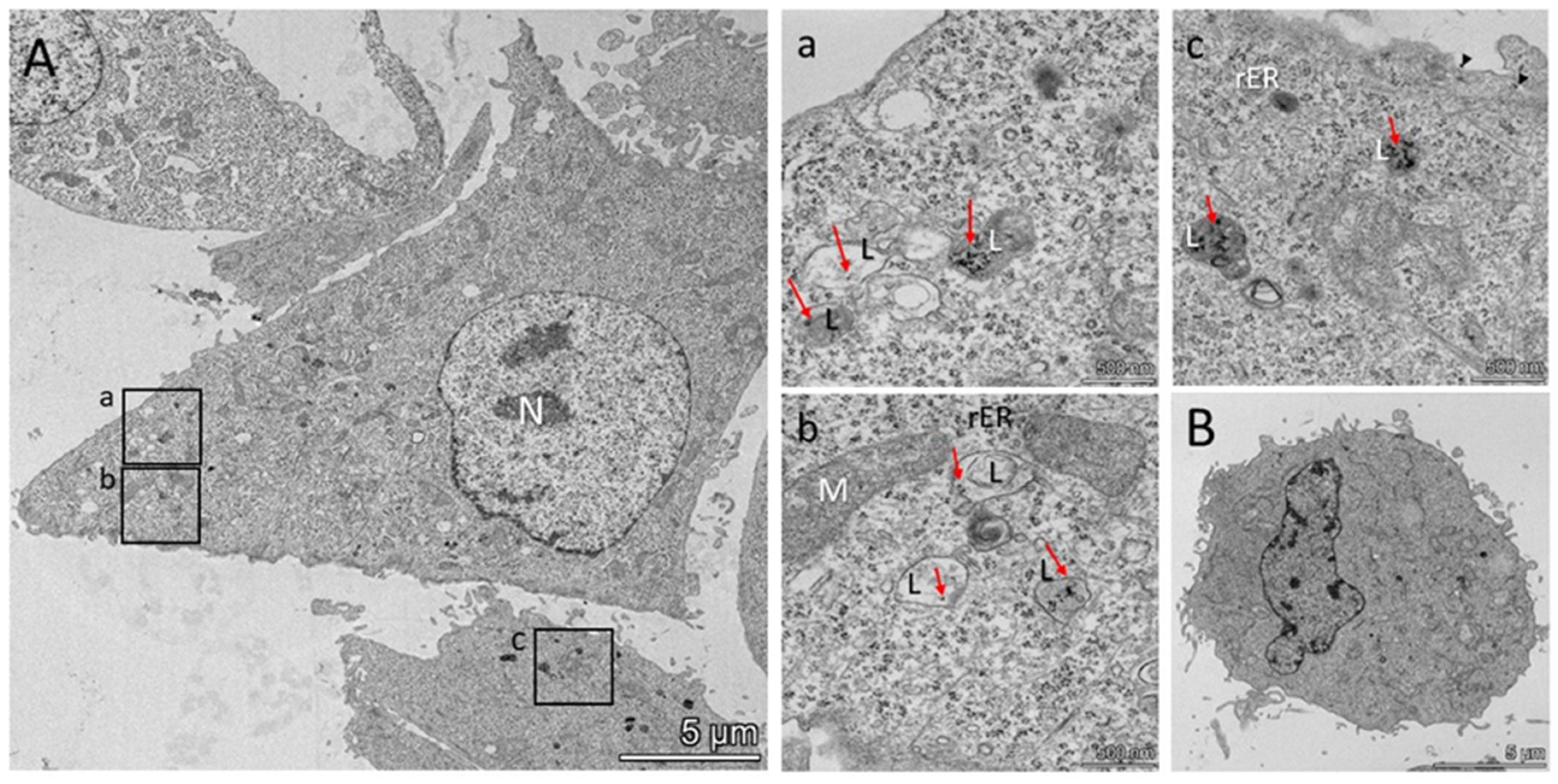
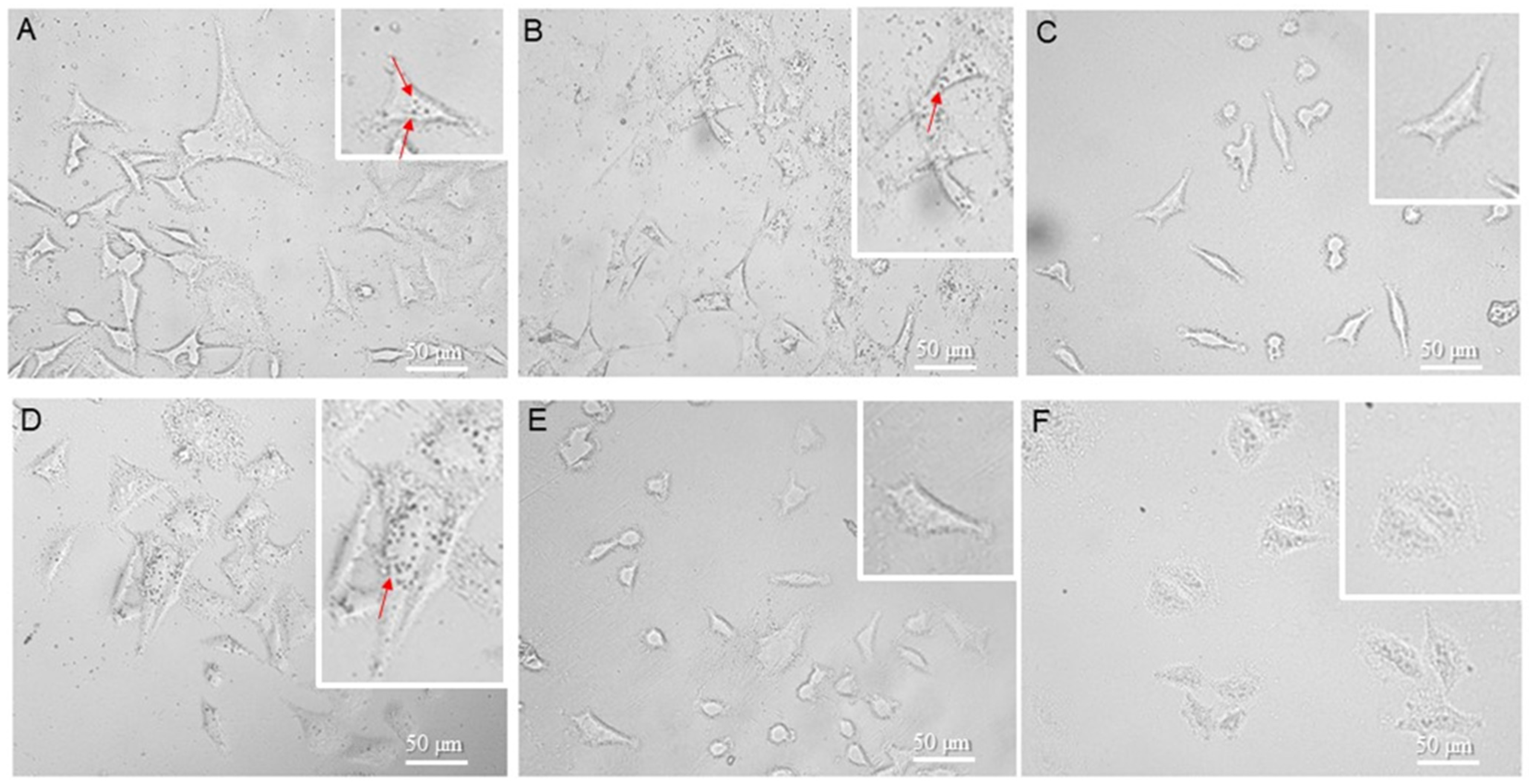
3.3. FeBNPs’ Interaction with HeLa Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jin, W.H.; Seldon, C.; Butkus, M.; Sauerwein, W.; Giap, H.B. A Review of Boron Neutron Capture Therapy: Its History and Current Challenges. Int. J. Part. Ther. 2022, 9, 71–82. [Google Scholar] [CrossRef]
- Barth, R.F.; Mi, P.; Yang, W. Boron Delivery Agents for Neutron Capture Therapy of Cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef]
- Monti Hughes, A.; Hu, N. Optimizing Boron Neutron Capture Therapy (BNCT) to Treat Cancer: An Updated Review on the Latest Developments on Boron Compounds and Strategies. Cancers 2023, 15, 4091. [Google Scholar] [CrossRef]
- Dymova, M.A.; Taskaev, S.Y.; Richter, V.A.; Kuligina, E.V. Boron Neutron Capture Therapy: Current Status and Future Perspectives. Cancer Commun. 2020, 40, 406–421. [Google Scholar] [CrossRef]
- Xuan, S.; Vicente, M.G.H. Recent Advances in Boron Delivery Agents for Boron Neutron Capture Therapy (BNCT); Wiley Online Library: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Zhang, Z.; Chong, Y.; Liu, Y.; Pan, J.; Huang, C.; Sun, Q.; Liu, Z.; Zhu, X.; Shao, Y.; Jin, C.; et al. A Review of Planned, Ongoing Clinical Studies and Recent Development of BNCT in Mainland of China. Cancers 2023, 15, 4060. [Google Scholar] [CrossRef]
- Moss, R.L. Critical Review, with an Optimistic Outlook, on Boron Neutron Capture Therapy (BNCT). Appl. Radiat. Isot. 2014, 88, 2–11. [Google Scholar] [CrossRef]
- Malouff, T.D.; Seneviratne, D.S.; Ebner, D.K.; Stross, W.C.; Waddle, M.R.; Trifiletti, D.M.; Krishnan, S. Boron Neutron Capture Therapy: A Review of Clinical Applications. Front. Oncol. 2021, 11, 601820. [Google Scholar] [CrossRef]
- Suzuki, M.; Kato, I.; Aihara, T.; Hiratsuka, J.; Yoshimura, K.; Niimi, M.; Kimura, Y.; Ariyoshi, Y.; Haginomori, S.I.; Sakurai, Y.; et al. Boron Neutron Capture Therapy Outcomes for Advanced or Recurrent Head and Neck Cancer. J. Radiat. Res. 2014, 55, 146–153. [Google Scholar] [CrossRef]
- Tsuji, T.; Yoshitomi, H.; Ishikawa, Y.; Koshizaki, N.; Suzuki, M.; Usukura, J. A Method to Selectively Internalise Submicrometer Boron Carbide Particles into Cancer Cells Using Surface Transferrin Conjugation for Developing a New Boron Neutron Capture Therapy Agent. J. Exp. Nanosci. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Takai, S.; Wanibuchi, M.; Kawabata, S.; Takeuchi, K.; Sakurai, Y.; Suzuki, M.; Ono, K.; Miyatake, S.I. Reactor-Based Boron Neutron Capture Therapy for 44 Cases of Recurrent and Refractory High-Grade Meningiomas with Long-Term Follow-Up. Neuro-Oncol. 2022, 24, 90–98. [Google Scholar] [CrossRef]
- Blue, T.E.; Yanch, J.C. Accelerator-based epithermal neutron sources for Boron Neutron Capture Therapy of brain tumors. J. Neuro-Oncol. 2003, 62, 19–31. [Google Scholar] [CrossRef]
- Oloo, S.O.; Smith, K.M.; Vicente, M.D.G.H. Multi-Functional Boron-Delivery Agents for Boron Neutron Capture Therapy of Cancers. Cancers 2023, 15, 3277. [Google Scholar] [CrossRef] [PubMed]
- Capoulat, M.E.; Kreiner, A.J. Induced radioactivity in AB-BNCT: An analysis of the different facilities worldwide. Front. Nucl. Eng. 2023, 2, 1275396. [Google Scholar] [CrossRef]
- Cartelli, D.E.; Capoulat, M.E.; Baldo, M.; Sandin Suarez, J.C.; Igarzabal, M.; del Grosso, M.F.; Valda, A.A.; Canepa, N.; Minsky, D.M.; Conti, G.; et al. Status of low-energy accelerator-based BNCT worldwide and in Argentina. Appl. Radiat. Isot. 2020, 166, 109315. [Google Scholar] [CrossRef]
- Bortolussi, S.; Postuma, I.; Protti, N.; Provenzano, L.; Ferrari, C.; Cansolino, L.; Dionigi, P.; Galasso, O.; Gasparini, G.; Altieri, S.; et al. Understanding the Potentiality of Accelerator Based-Boron Neutron Capture Therapy for Osteosarcoma: Dosimetry Assessment Based on the Reported Clinical Experience. Radiat. Oncol. 2017, 12, 130. [Google Scholar] [CrossRef]
- Kreiner, A.J.; Bergueiro, J.; Cartelli, D.; Baldo, M.; Castell, W.; Asoia, J.G.; Padulo, J.; Suárez Sandín, J.C.; Igarzabal, M.; Erhardt, J.; et al. Present Status of Accelerator-Based BNCT. Rep. Pract. Oncol. Radiother. 2016, 21, 95–101. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Cheng Yan, K.; Maguire, J.A.; Hosmane, N.S. Recent Developments in Boron Neutron Capture Therapy (BNCT) Driven by Nanotechnology. Curr. Chem. Biol. 2008, 1, 141–149. [Google Scholar] [CrossRef]
- Beck-Sickinger, A.G.; Becker, D.P.; Chepurna, O.; Das, B.; Flieger, S.; Hey-Hawkins, E.; Hosomanie, N.; Jalisatgi, S.S.; Nakamura, H.; Patil, R.; et al. New Boron Delivery Agents. Cancer Biother. Radiopharm. 2023, 38, 160–172. Available online: https://pubmed.ncbi.nlm.nih.gov/36350709/ (accessed on 18 March 2024). [CrossRef] [PubMed]
- Zhang, X.; Lin, Y.; Hosmane, N.S.; Zhu, Y. Nanostructured Boron Agents for Boron Neutron Capture Therapy: A Review of Recent Patents. Med. Rev. 2023, 3, 425–443. [Google Scholar] [CrossRef]
- Singh, B.; Kaur, G.; Singh, P.; Singh, K.; Kumar, B.; Vij, A.; Kumar, M.; Bala, R.; Meena, R.; Singh, A.; et al. Nanostructured Boron Nitride with High Water Dispersibility for Boron Neutron Capture Therapy. Sci. Rep. 2016, 6, 35535. [Google Scholar] [CrossRef]
- Paul, W.; Sharma, C.P. Inorganic Nanoparticles for Targeted Drug Delivery; Elsevier Ltd.: Amsterdam, The Netherlands, 2019. [Google Scholar] [CrossRef]
- Gosset, D. Basic Properties of Boron Carbide. Compr. Nucl. Mater. (Second Ed.) 2020, 7, 539–553. [Google Scholar] [CrossRef]
- Tatiya, S.; Pandey, M.; Bhattacharya, S. Nanoparticles Containing Boron and Its Compounds—Synthesis and Applications: A Review. J. Micromanufacturing 2020, 3, 159–173. [Google Scholar] [CrossRef]
- Yoshie Ishikawa, T.I. Boron Carbide Particle as a Boron Compound for Boron Neutron Capture Therapy. J. Nucl. Med. Radiat. Ther. 2014, 5, 2–6. [Google Scholar] [CrossRef]
- Mortensen, M.W.; Sørensen, P.G.; Björkdahl, O.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Preparation and Characterization of Boron Carbide Nanoparticles for Use as a Novel Agent in T Cell-Guided Boron Neutron Capture Therapy. Appl. Radiat. Isot. 2006, 64, 315–324. [Google Scholar] [CrossRef]
- Mortensen, M.W.; Björkdahl, O.; Sørensen, P.G.; Hansen, T.; Jensen, M.R.; Gundersen, H.J.G.; Bjørnholm, T. Functionalization and Cellular Uptake of Boron Carbide Naraoparticles. The First Step toward T Cell-Guided Boron Neutron Capture Therapy. Bioconjug. Chem. 2006, 17, 284–290. [Google Scholar] [CrossRef]
- Vitali, A.; Demichelis, M.P.; Di Martino, G.; Postuma, I.; Bortolussi, S.; Falqui, A.; Milanese, C.; Ferrara, C.; Sommi, P.; Anselmi-Tamburini, U. Synthesis and Characterization of Gd-Functionalized B4C Nanoparticles for BNCT Applications. Life 2023, 13, 429. [Google Scholar] [CrossRef]
- Kozień, D.; Żeliszewska, P.; Szermer-Olearnik, B.; Adamczyk, Z.; Wróblewska, A.; Szczygieł, A.; Węgierek-Ciura, K.; Mierzejewska, J.; Pajtasz-Piasecka, E.; Tokarski, T.; et al. Synthesis and Characterization of Boron Carbide Nanoparticles as Potential Boron-Rich Therapeutic Carriers. Materials 2023, 16, 6534. [Google Scholar] [CrossRef] [PubMed]
- Wallyn, J.; Anton, N.; Vandamme, T.F. Synthesis, Principles, and Properties of Magnetite Nanoparticles for in Vivo Imaging Applications—A Review. Pharmaceutics 2019, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Jeon, M.; Halbert, M.V.; Stephen, Z.R.; Zhang, M. Iron Oxide Nanoparticles as T1 Contrast Agents for Magnetic Resonance Imaging: Fundamentals, Challenges, Applications, and Prospectives. Adv. Mater. 2021, 33, 1906539. [Google Scholar] [CrossRef] [PubMed]
- Stephen, Z.R.; Kievit, F.M.; Zhang, M. Magnetite Nanoparticles for Medical MR Imaging. Mater. Today 2011, 14, 330–338. [Google Scholar] [CrossRef]
- Thakor, A.S.; Jokerst, J.V.; Ghanouni, P.; Campbell, J.L.; Mittra, E.; Gambhir, S.S. Clinically Approved Nanoparticle Imaging Agents. J. Nucl. Med. 2016, 57, 1833–1837. [Google Scholar] [CrossRef]
- Revia, R.A.; Zhang, M. Magnetite Nanoparticles for Cancer Diagnosis, Treatment, and Treatment Monitoring: Recent Advances. Mater. Today 2016, 19, 157–168. [Google Scholar] [CrossRef]
- Aladesuyi, O.A.; Oluwafemi, S.O. The role of magnetic nanoparticles in cancer management. Nano-Struct. Nano-Objects 2023, 36, 101053. [Google Scholar] [CrossRef]
- Chengyin, F.; Ravindra, N.M. Magnetic iron oxide nanoparticles: Synthesis and applications. Bioinspired Biomim. Nanobiomaterials 2012, 1, 229–244. [Google Scholar] [CrossRef]
- Santra, S.; Kaittanis, C.; Grimm, J.; Perez, J.M. Drug/Dye-Loaded, Multifunctional Iron Oxide Nanoparticles for Combined Targeted Cancer Therapy and Dual Optical/Magnetic Resonance Imaging. Small 2009, 5, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium Salts and Formazan Products in Cell Biology: Viability Assessment, Fluorescence Imaging, and Labeling Perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef]
- Tan, X.; Zhou, R.; Feng, Y.; Liang, T. In-Depth Method Investigation for Determination of Boron in Silicate Samples Using an Improved Boron–Mannitol Complex Digestion Method by Inductively Coupled Plasma Mass Spectrometry. Molecules 2023, 28, 441. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.N.; Brown, P.H. Boron Determination—A Review of Analytical Methods. Microchem. J. 1997, 304, 285–304. [Google Scholar] [CrossRef]
- Portu, A.; Rossini, A.E.; Thorp, S.I.; Curotto, P.; Pozzi, E.C.C.; Granell, P.; Golmar, F.; Cabrini, R.L.; Martin, G.S. Simultaneous Observation of Cells and Nuclear Tracks from the Boron Neutron Capture Reaction by UV-C Sensitization of Polycarbonate. Microsc. Microanal. 2015, 21, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Gadan, M.A.; Lloyd, R.; Saint Martin, G.; Olivera, M.S.; Policastro, L.; Portu, A.M. Neutron Autoradiography Combined with UV-C Sensitization: Toward the Intracellular Localization of Boron. Microsc. Microanal. 2019, 25, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Portu, A.M.; Espain, M.S.; Thorp, S.I.; Trivillin, V.A.; Curotto, P.; Monti Hughes, A.; Pozzi, E.C.C.; Garabalino, M.A.; Palmieri, M.A.; Granell, P.N.; et al. Enhanced Resolution of Neutron Autoradiography with UV-C Sensitization to Study Boron Microdistribution in Animal Models. Life 2023, 13, 1578. [Google Scholar] [CrossRef] [PubMed]
- Sezer, A.O.; Brand, J.I. Chemical Vapor Deposition of Boron Carbide Chemical Vapor Deposition of Boron Carbide. Mater. Sci. Eng. 2019, 79, 191–202. [Google Scholar] [CrossRef]
- Suri, A.K.; Subramanian, C.; Sonber, J.K.; Ch Murthy, T.S.R. Synthesis and Consolidation of Boron Carbide: A Review. Int. Mater. Rev. 2010, 55, 4–40. [Google Scholar] [CrossRef]
- Kiliçarslan, A.; Toptan, F.; Kerti, I.; Piskin, S. Oxidation of Boron Carbide Particles at Low Temperatures. Mater. Lett. 2014, 128, 224–226. [Google Scholar] [CrossRef]
- Kozién, D.; Jelén, P.; Stepién, J.; Olejniczak, Z.; Sitarz, M.; Pedzich, Z. Surface Properties and Morphology of Boron Carbide Nanopowders Obtained by Lyophilization of Saccharide Precursors. Materials 2021, 14, 3419. [Google Scholar] [CrossRef]
- Rotaru, A. Thermal and Kinetic Study of Hexagonal Boric Acid versus Triclinic Boric Acid in Air Flow. J. Therm. Anal. Calorim. 2017, 127, 755–763. [Google Scholar] [CrossRef]
- Jain, A.; Anthonysamy, S. Oxidation of Boron Carbide powder. J. Therm. Anal. Calorim. 2015, 122, 645–652. [Google Scholar] [CrossRef]
- Bin-Dahman, O.A.; Jose, J.; Al-Harthi, M.A. Compatibility of Poly(Acrylic Acid)/Starch Blends. Starch/Staerke 2015, 67, 1061–1069. [Google Scholar] [CrossRef]
- Sousa de Almeida, M.; Susnik, E.; Drasler, B.; Taladriz-Blanco, P.; Petri-Fink, A.; Rothen-Rutishauser, B. Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine. Chem. Soc. Rev. 2021, 50, 5397. [Google Scholar] [CrossRef]
- Saint Martin, G.; Portu, A.M.; Ibarra, M.L.; Alurralde, M. UV-C Radiation Effect on Nuclear Tracks of Different Ions in Polycarbonate. Radiat. Phys. Chem. 2020, 173, 108936. [Google Scholar] [CrossRef]
- Portu, A.; Postuma, I.; Gadan, M.A.; Saint Martin, G.; Olivera, M.S.; Altieri, S.; Protti, N.; Bortolussi, S. Reprint of Inter-Comparison of Boron Concentration Measurements at INFN-University of Pavia (Italy) and CNEA (Argentina). Appl. Radiat. Isot. 2015, 106, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Langenbacher, R.E.; Horoszko, C.P.; Kim, M.; Heller, D. Hematoxylin Nuclear Stain Reports Oxidative Stress via Near-Infrared Emission. ACS Chem. Biol. 2023, 18, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- International Atomic Energy Agency. Advances in Boron Neutron Capture Therapy; IAEAL 23-01601 (paperback: Alk. paper); International Atomic Energy Agency: Vienna, Austria, 2023; ISBN 978-92-0-132723-9/978-92-0-132623-2. [Google Scholar]
- Mechetin, G.V.; Zharkov, D.O. DNA Damage Response and Repair in Boron Neutron Capture Therapy. Genes 2023, 14, 127. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demichelis, M.P.; Portu, A.M.; Gadan, M.A.; Vitali, A.; Forlingieri, V.; Bortolussi, S.; Postuma, I.; Falqui, A.; Vezzoli, E.; Milanese, C.; et al. Synthesis and Characterization of B4C-Based Multifunctional Nanoparticles for Boron Neutron Capture Therapy Applications. Appl. Nano 2024, 5, 33-47. https://doi.org/10.3390/applnano5020004
Demichelis MP, Portu AM, Gadan MA, Vitali A, Forlingieri V, Bortolussi S, Postuma I, Falqui A, Vezzoli E, Milanese C, et al. Synthesis and Characterization of B4C-Based Multifunctional Nanoparticles for Boron Neutron Capture Therapy Applications. Applied Nano. 2024; 5(2):33-47. https://doi.org/10.3390/applnano5020004
Chicago/Turabian StyleDemichelis, Maria Paola, Agustina Mariana Portu, Mario Alberto Gadan, Agostina Vitali, Valentina Forlingieri, Silva Bortolussi, Ian Postuma, Andrea Falqui, Elena Vezzoli, Chiara Milanese, and et al. 2024. "Synthesis and Characterization of B4C-Based Multifunctional Nanoparticles for Boron Neutron Capture Therapy Applications" Applied Nano 5, no. 2: 33-47. https://doi.org/10.3390/applnano5020004