

Enhancing NO Uptake in Metal-Organic Frameworks via Linker Functionalization. A Multi-Scale Theoretical Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodology
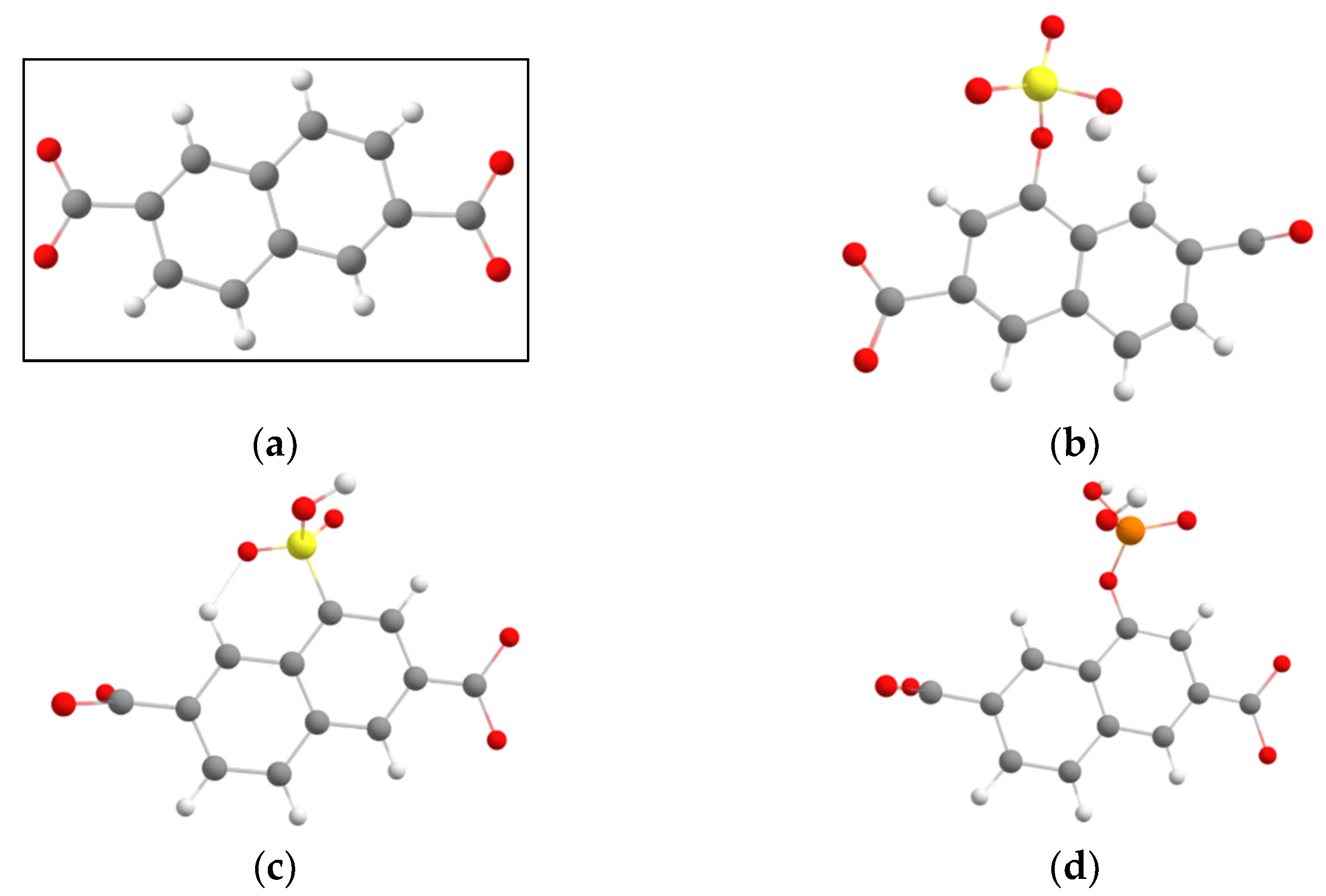
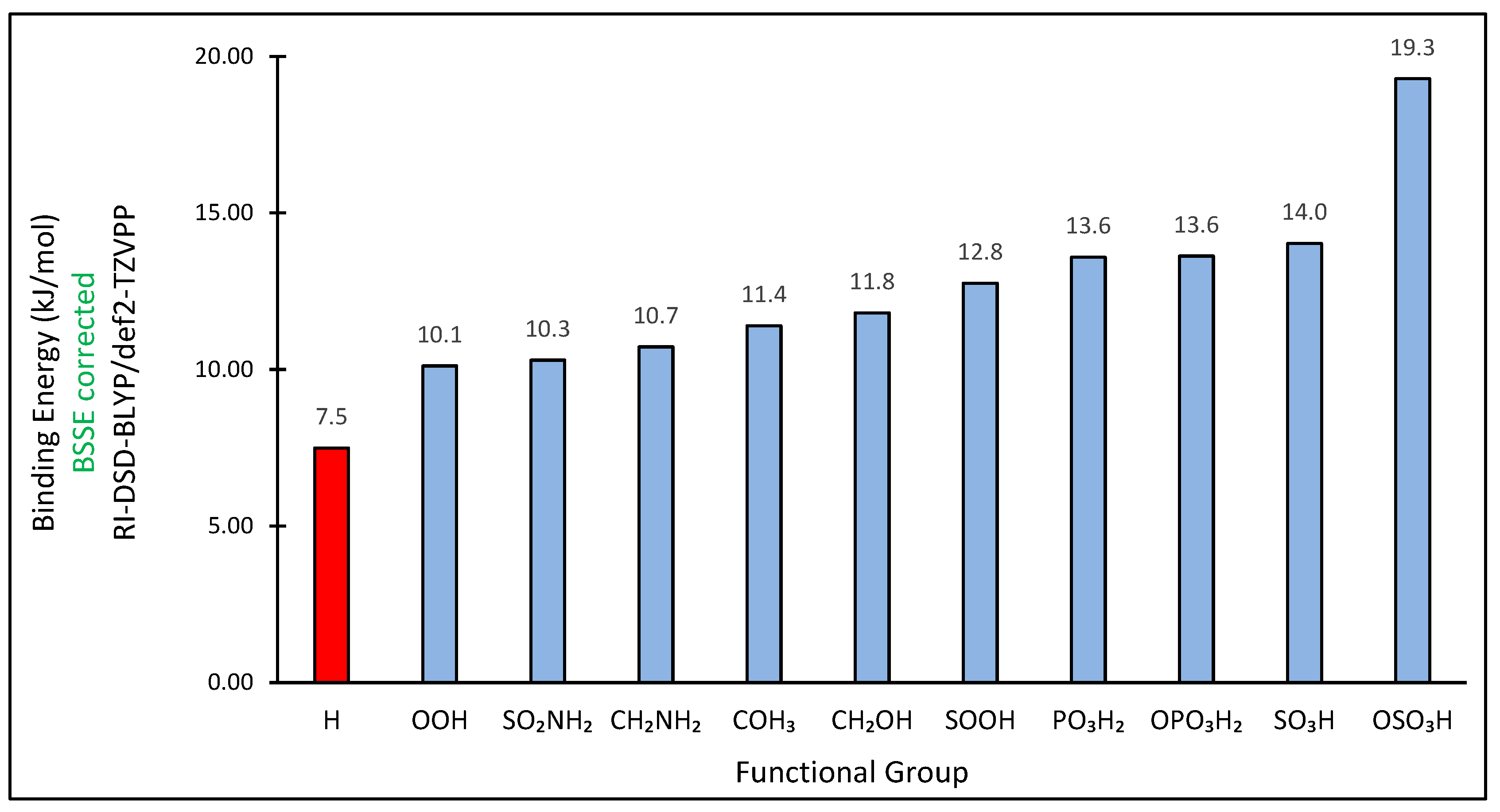
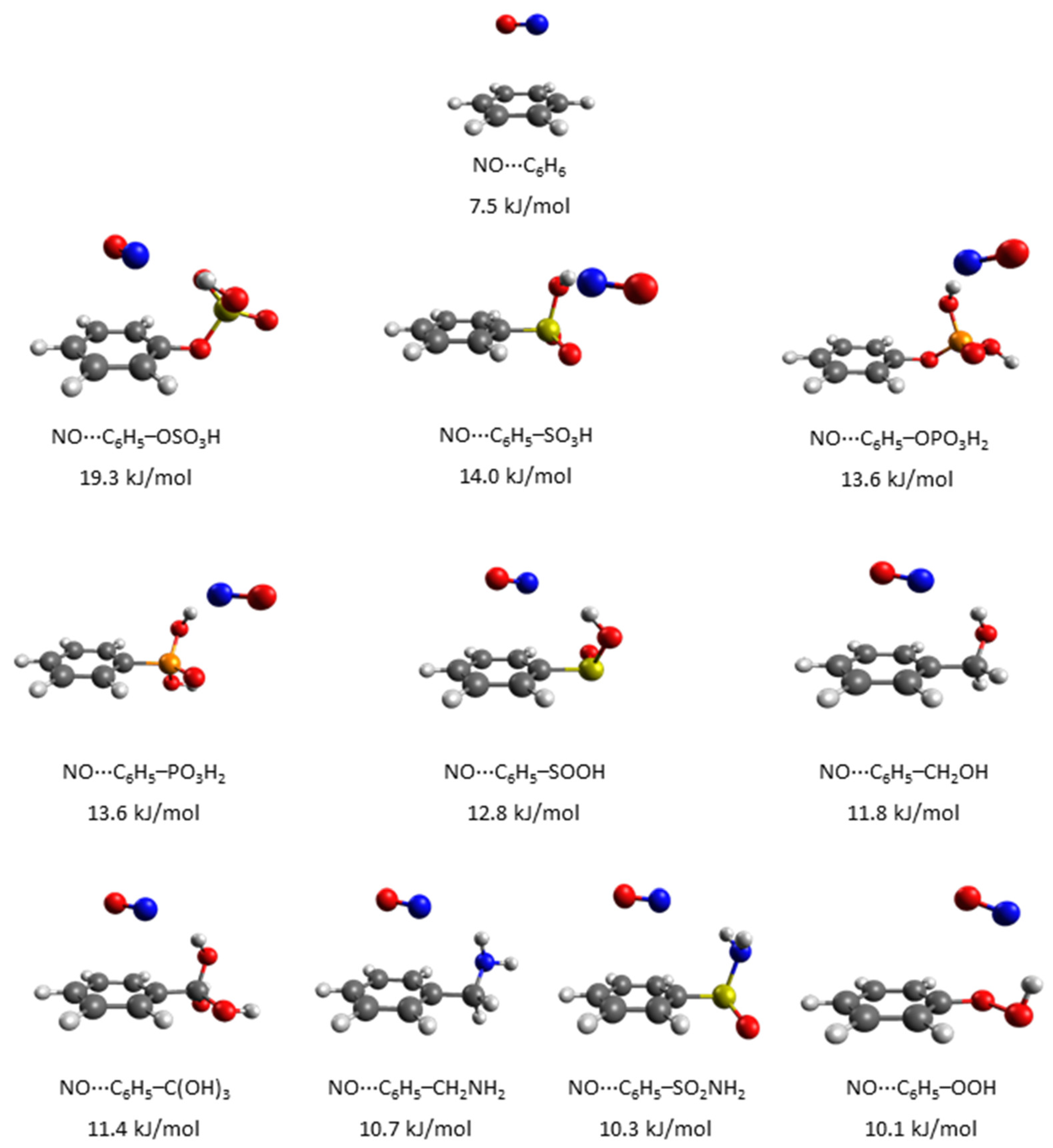
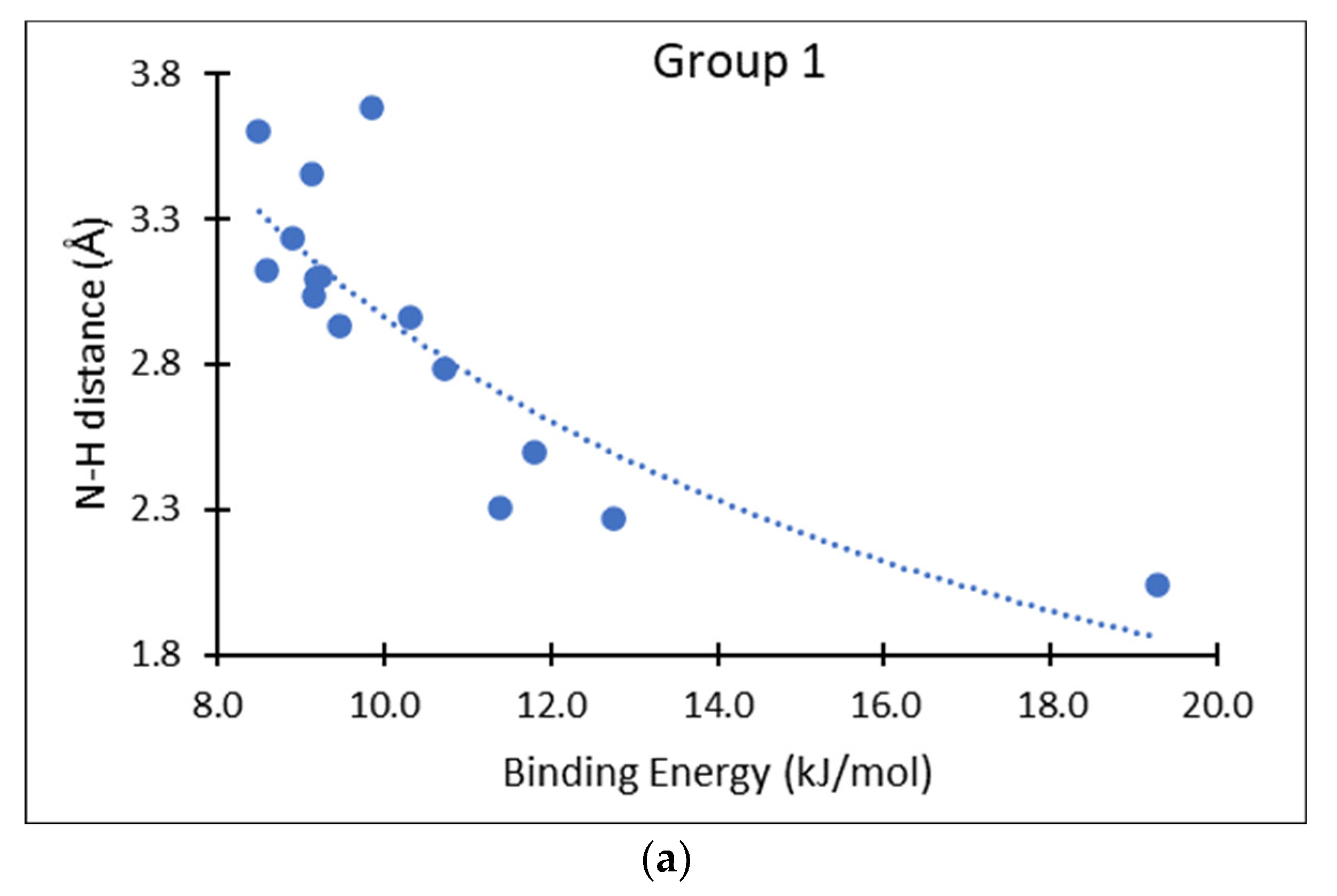
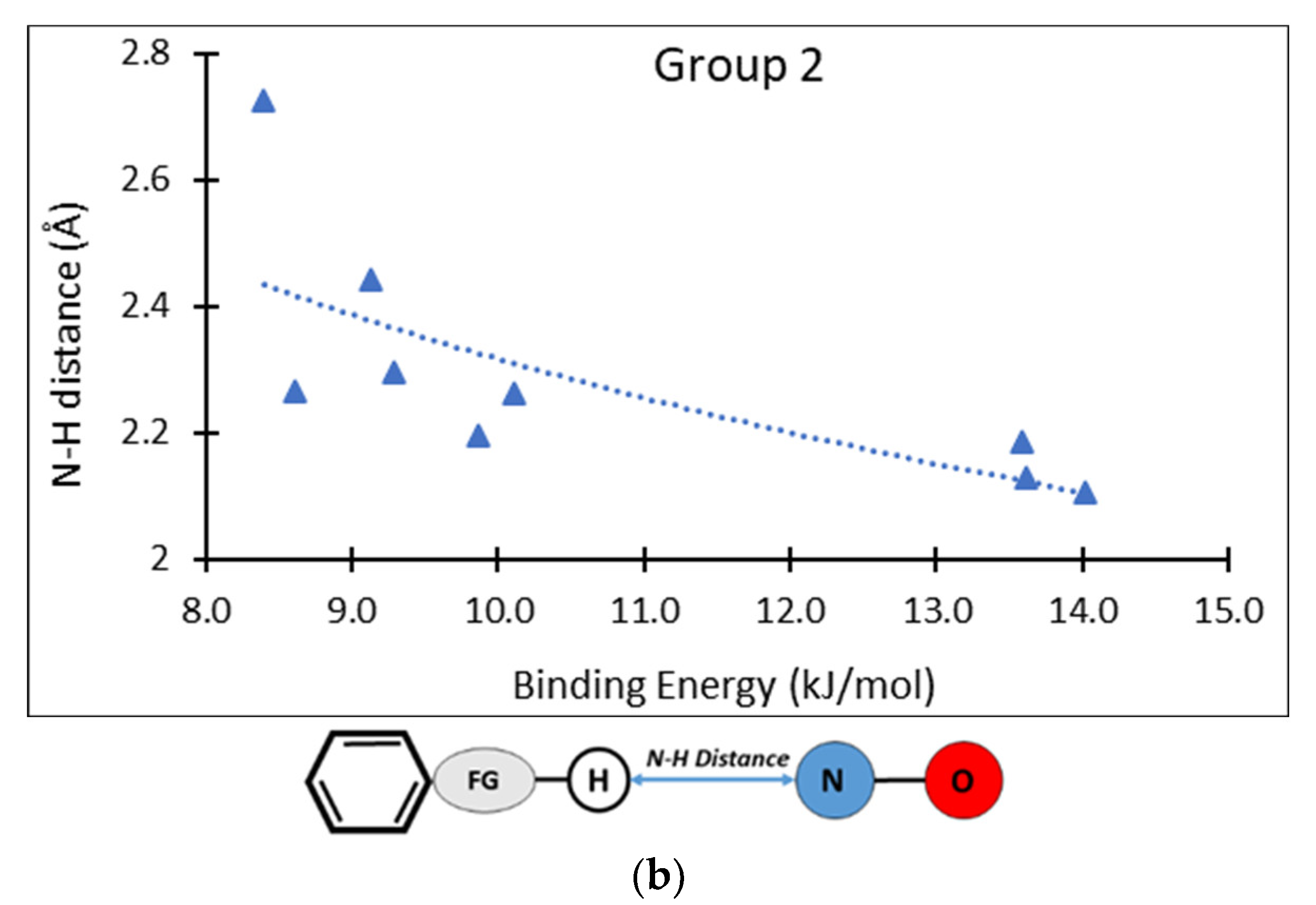
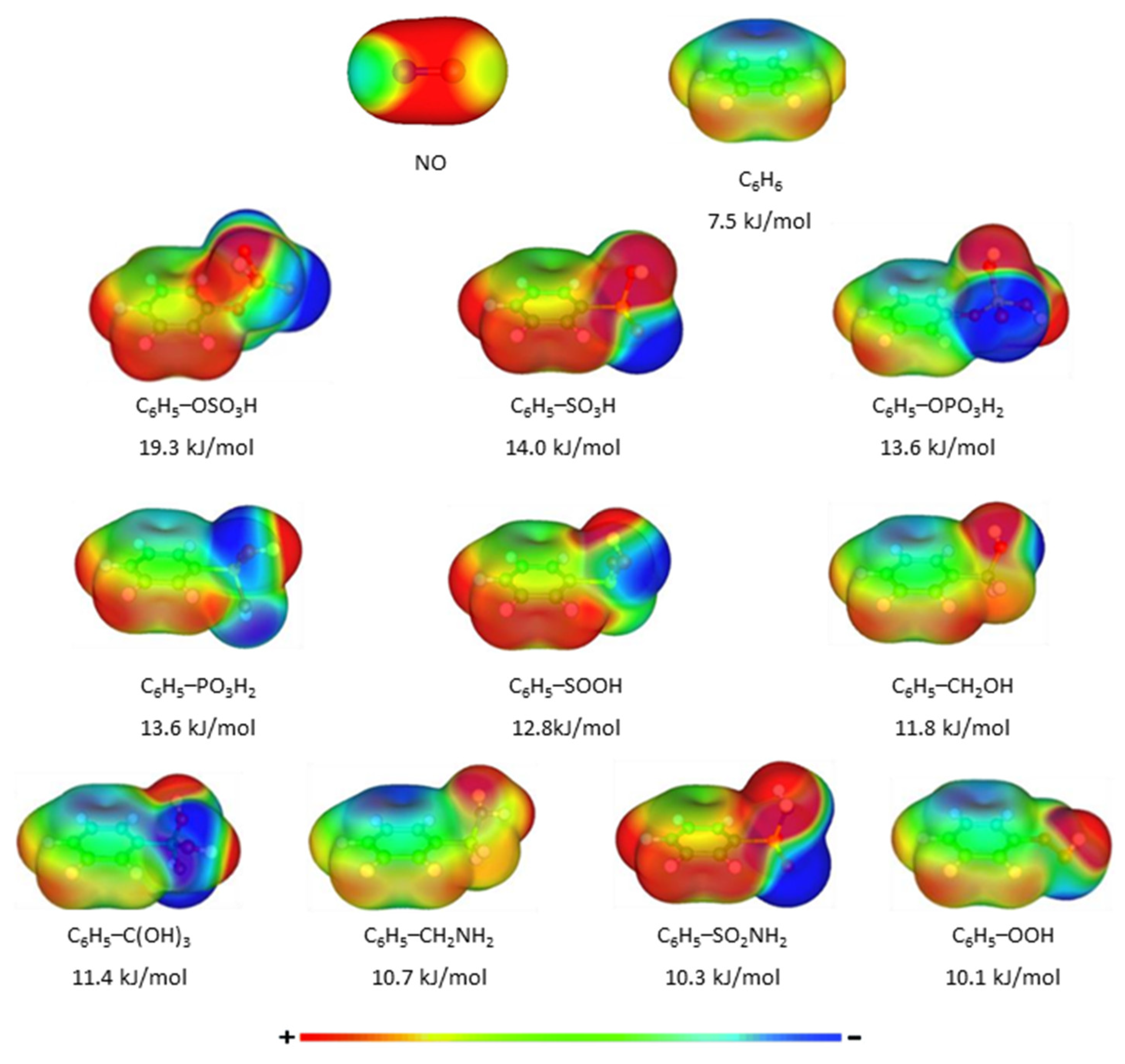
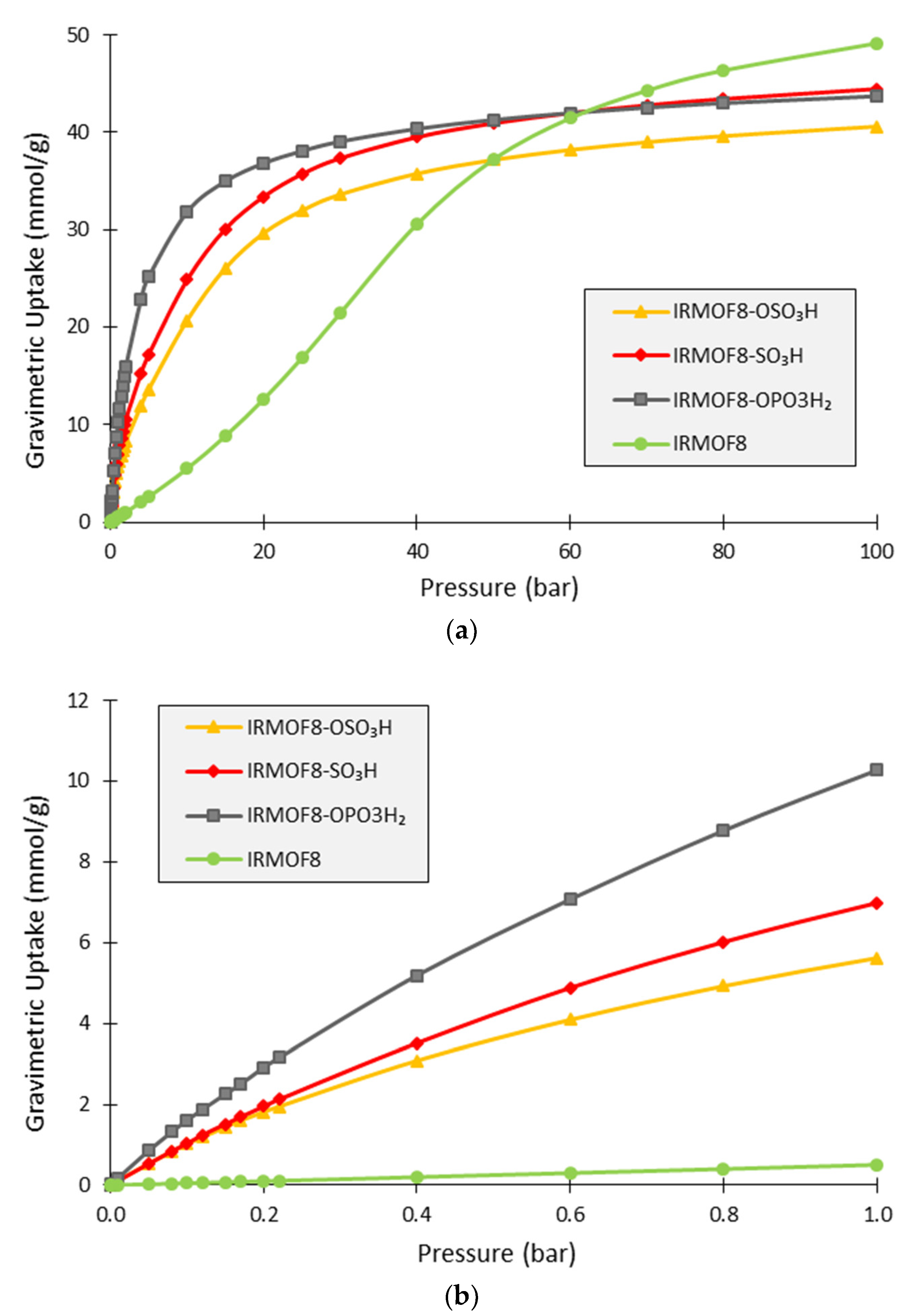
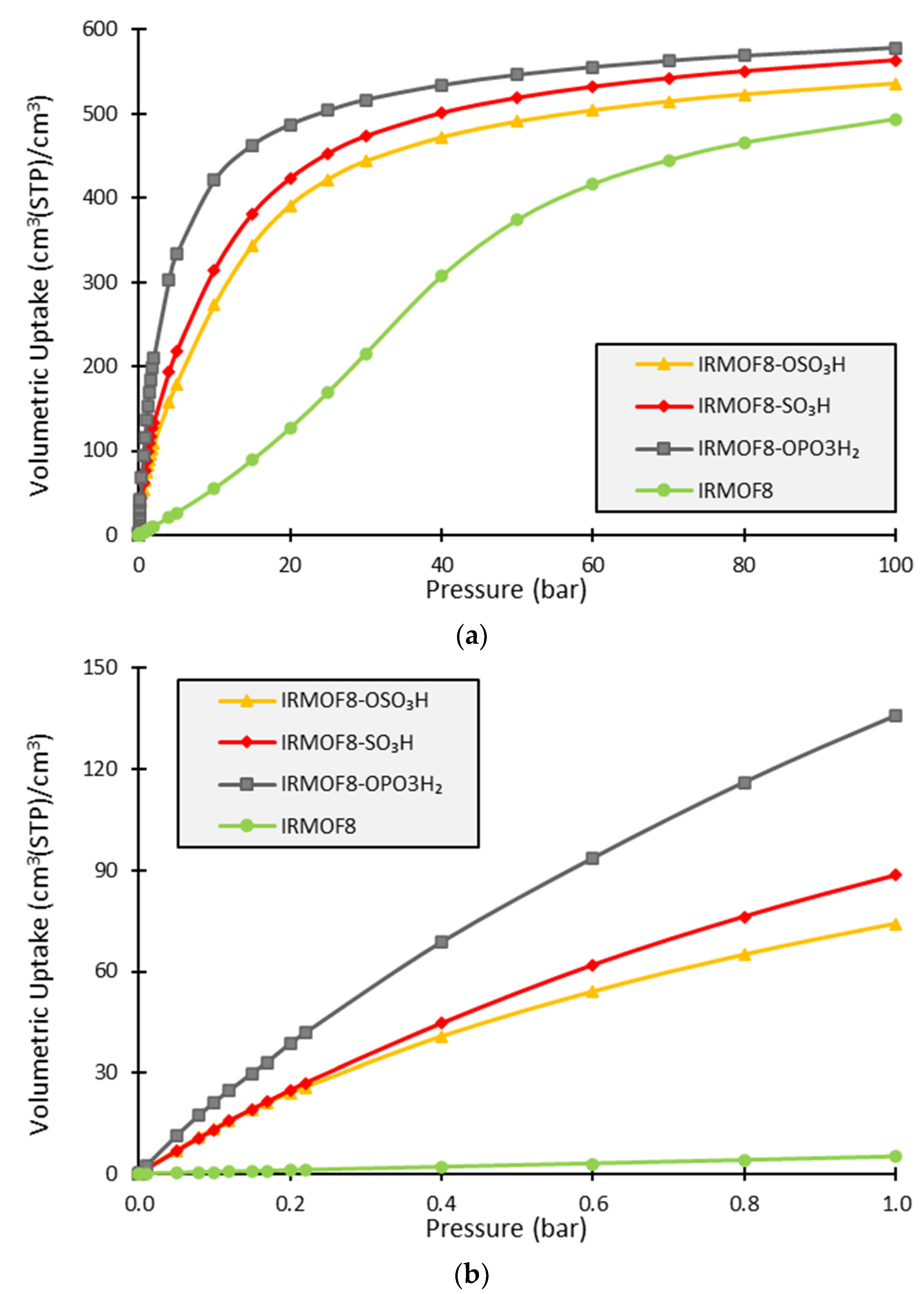
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hill, S.C.; Douglas Smoot, L. Modeling of nitrogen oxides formation and destruction in combustion systems. Prog. Energy Combust. Sci. 2000, 26, 417–458. [Google Scholar] [CrossRef]
- European Environment Agency. Nitrogen Oxides (NOx) Emissions. Available online: https://www.eea.europa.eu/data-and-maps/indicators/eea-32-nitrogen-oxides-nox-emissions-1/assessment.2010-08-19.0140149032-3 (accessed on 12 July 2022).
- European Environment Agency. Air Pollution Sources. Available online: https://www.eea.europa.eu/themes/air/air-pollution-sources-1 (accessed on 6 June 2022).
- Mannucci, P.M.; Harari, S.; Martinelli, I.; Franchini, M. Effects on health of air pollution: A narrative review. Intern. Emerg. Med. 2015, 10, 657–662. [Google Scholar] [CrossRef]
- Air Pollution. Available online: https://www.who.int/data/gho/data/themes/theme-details/GHO/air-pollution (accessed on 6 June 2022).
- World Bank. The Global Health Cost of PM2.5 Air Pollution: A Case for Action Beyond 2021; World Bank: Washington, DC, USA, 2022; ISBN 978-1-4648-1816-5. [Google Scholar]
- US EPA. What Is Acid Rain? Available online: https://www.epa.gov/acidrain/what-acid-rain (accessed on 6 June 2022).
- US EPA. Effects of Acid Rain. Available online: https://www.epa.gov/acidrain/effects-acid-rain (accessed on 6 June 2022).
- Yaghi, O.M.; Li, G.; Li, H. Selective binding and removal of guests in a microporous metal–organic framework. Nature 1995, 378, 703–706. [Google Scholar] [CrossRef]
- Batten, S.R.; Robson, R. Interpenetrating Nets: Ordered, Periodic Entanglement. Angew. Chem. Int. Ed. Engl. 1998, 37, 1460–1494. [Google Scholar] [CrossRef]
- Férey, G. Some suggested perspectives for multifunctional hybrid porous solids. Dalton Trans. 2009, 23, 4400–4415. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.A.; Wöll, C. Functionalized Coordination Space in Metal–Organic Frameworks. Angew. Chem. Int. Ed. 2008, 47, 8164–8168. [Google Scholar] [CrossRef]
- Kitagawa, S.; Matsuda, R. Chemistry of coordination space of porous coordination polymers. Coord. Chem. Rev. 2007, 251, 2490–2509. [Google Scholar] [CrossRef]
- Kepert, C.J. Advanced functional properties in nanoporous coordination framework materials. Chem. Commun. 2006, 7, 695–700. [Google Scholar] [CrossRef]
- Mueller, U.; Schubert, M.; Teich, F.; Puetter, H.; Schierle-Arndt, K.; Pastré, J. Metal–organic frameworks—Prospective industrial applications. J. Mater. Chem. 2006, 16, 626–636. [Google Scholar] [CrossRef]
- Lin, J.; Ho, W.; Qin, X.; Leung, C.; Au, V.K.; Lee, S. Metal–Organic Frameworks for NOx Adsorption and Their Applications in Separation, Sensing, Catalysis, and Biology. Small 2022, 18, 2105484. [Google Scholar] [CrossRef]
- Xiao, B.; Wheatley, P.S.; Zhao, X.; Fletcher, A.J.; Fox, S.; Rossi, A.G.; Megson, I.L.; Bordiga, S.; Regli, L.; Thomas, K.M.; et al. High-Capacity Hydrogen and Nitric Oxide Adsorption and Storage in a Metal−Organic Framework. J. Am. Chem. Soc. 2007, 129, 1203–1209. [Google Scholar] [CrossRef]
- McKinlay, A.C.; Eubank, J.F.; Wuttke, S.; Xiao, B.; Wheatley, P.S.; Bazin, P.; Lavalley, J.-C.; Daturi, M.; Vimont, A.; De Weireld, G.; et al. Nitric Oxide Adsorption and Delivery in Flexible MIL-88(Fe) Metal–Organic Frameworks. Chem. Mater. 2013, 25, 1592–1599. [Google Scholar] [CrossRef]
- Livas, C.G.; Tylianakis, E.; Froudakis, G.E. Enhancing of CO Uptake in Metal–Organic Frameworks by Linker Functionalization: A Multi-Scale Theoretical Study. Chemistry 2022, 4, 43. [Google Scholar] [CrossRef]
- Raptis, D.; Livas, C.; Stavroglou, G.; Giappa, R.M.; Tylianakis, E.; Stergiannakos, T.; Froudakis, G.E. Surface Modification Strategy for Enhanced NO2 Capture in Metal–Organic Frameworks. Molecules 2022, 27, 3448. [Google Scholar] [CrossRef]
- Frysali, M.G.; Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Tuning the interaction strength and the adsorption of CO2 in metal organic frameworks by functionalization of the organic linkers. Microporous Mesoporous Mater. 2016, 227, 144–151. [Google Scholar] [CrossRef]
- Giappa, R.M.; Tylianakis, E.; Di Gennaro, M.; Gkagkas, K.; Froudakis, G.E. A combination of multi-scale calculations with machine learning for investigating hydrogen storage in metal organic frameworks. Int. J. Hydrogen Energy 2021, 46, 27612–27621. [Google Scholar] [CrossRef]
- Stergiannakos, T.; Klontzas, E.; Tylianakis, E.; Froudakis, G.E. Enhancement of CO2 Adsorption in Magnesium Alkoxide IRMOF-10. J. Phys. Chem. C 2015, 119, 22001–22007. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Kozuch, S.; Gruzman, D.; Martin, J.M.L. DSD-BLYP: A General Purpose Double Hybrid Density Functional Including Spin Component Scaling and Dispersion Correction. J. Phys. Chem. C 2010, 114, 20801–20808. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy—Physical Chemistry Chemical Physics (RSC Publishing). Available online: https://pubs.rsc.org/en/content/articlelanding/2005/cp/b508541a (accessed on 7 June 2022).
- Skylaris, C.-K.; Gagliardi, L.; Handy, N.C.; Ioannou, A.G.; Spencer, S.; Willetts, A. On the resolution of identity Coulomb energy approximation in density functional theory. J. Mol. Struct. THEOCHEM 2000, 501–502, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Laaksonen, L. A graphics program for the analysis and display of molecular dynamics trajectories. J. Mol. Graph. 1992, 10, 33–34, 24. [Google Scholar] [CrossRef]
- Bergman, D.L.; Laaksonen, L.; Laaksonen, A. Visualization of solvation structures in liquid mixtures. J. Mol. Graph. Model 1997, 15, 301–306, 328–333. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simul. 2016, 42, 81–101. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.E.; Chapman, S. On the determination of molecular fields.—II. From the equation of state of a gas. Proc. R. Soc. Lond. A 1924, 106, 463–477. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ren, Y.; Tian, A.; Sun, H. COMPASS Force Field for 14 Inorganic Molecules, He, Ne, Ar, Kr, Xe, H2, O2, N2, NO, CO, CO2, NO2, CS2, and SO2, in Liquid Phases. J. Phys. Chem. B 2000, 104, 4951–4957. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Lorentz, H.A. Ueber die Anwendung des Satzes vom Virial in der kinetischen Theorie der Gase. Ann. Phys. 1881, 248, 127–136. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Livas, C.G.; Tylianakis, E.; Froudakis, G.E. Enhancing NO Uptake in Metal-Organic Frameworks via Linker Functionalization. A Multi-Scale Theoretical Study. Chemistry 2022, 4, 1300-1311. https://doi.org/10.3390/chemistry4040086
Livas CG, Tylianakis E, Froudakis GE. Enhancing NO Uptake in Metal-Organic Frameworks via Linker Functionalization. A Multi-Scale Theoretical Study. Chemistry. 2022; 4(4):1300-1311. https://doi.org/10.3390/chemistry4040086
Chicago/Turabian StyleLivas, Charalampos G., Emmanuel Tylianakis, and George E. Froudakis. 2022. "Enhancing NO Uptake in Metal-Organic Frameworks via Linker Functionalization. A Multi-Scale Theoretical Study" Chemistry 4, no. 4: 1300-1311. https://doi.org/10.3390/chemistry4040086