
Copper(II) NHC Catalyst for the Formation of Phenol from Arylboronic Acid

 ,
,  ,
,  , , and
, , and
Abstract
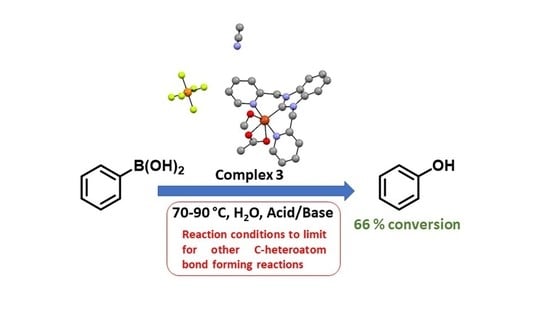
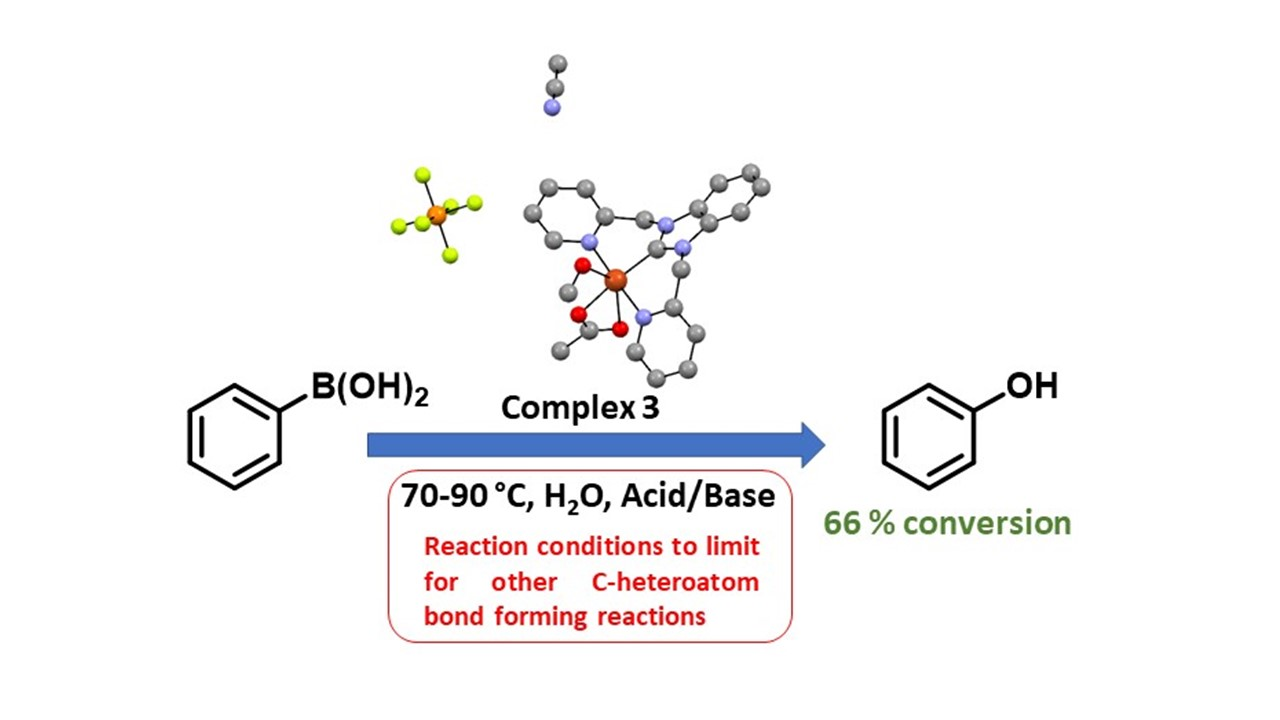
:
1. Introduction
2. Materials and Methods
2.1. Materials
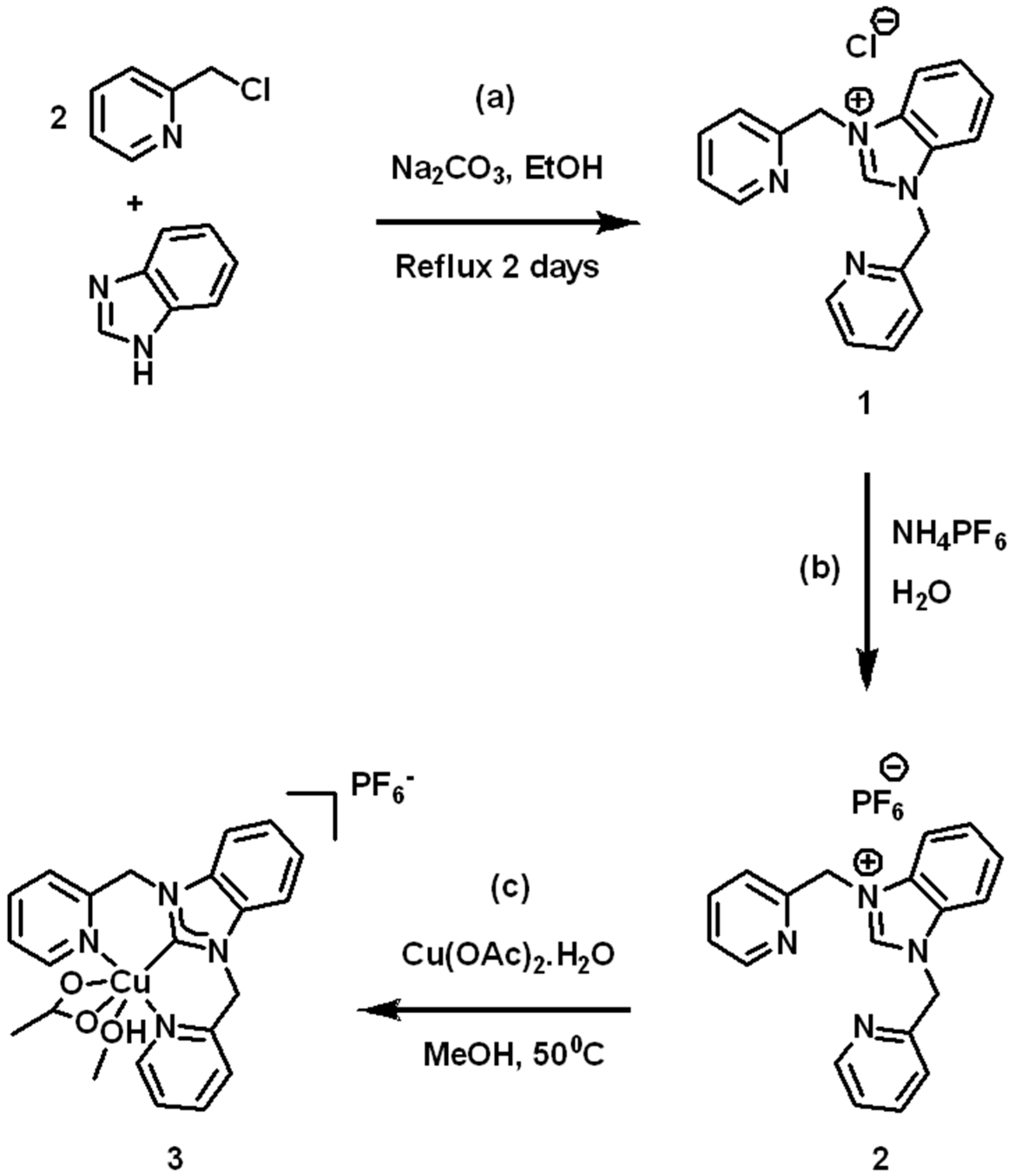
2.2. Synthesis of 1,3-bis(Pyridin-2-ylmethyl)-1H-benzo[d]imidazol-3-ium Chloride (1)
2.3. Synthesis of 1,3-bis(Pyridin-2-ylmethyl)-1H-benzo[d]imidazol-3-ium Hexafluorophosphate (2)
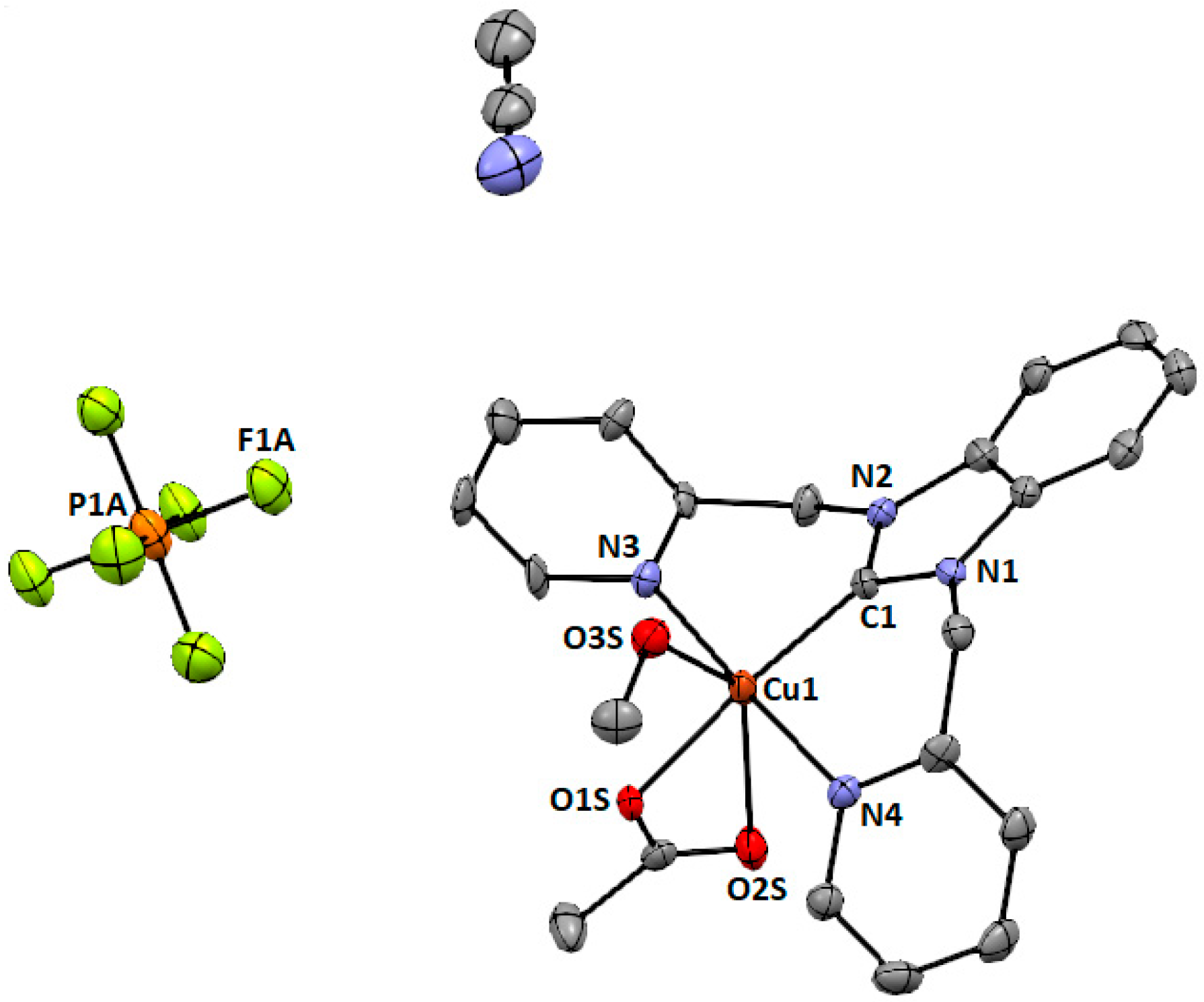
2.4. Synthesis of Copper(II) (1,3-bis(Pyridin-2-ylmethyl)-1H-benzo[d]imidazol-3-ium) Hexafluorophosphate (3)
2.5. General Procedure for the Copper-NHC-Catalyzed Formation of Phenol and Ethers
2.6. Experimental Methods
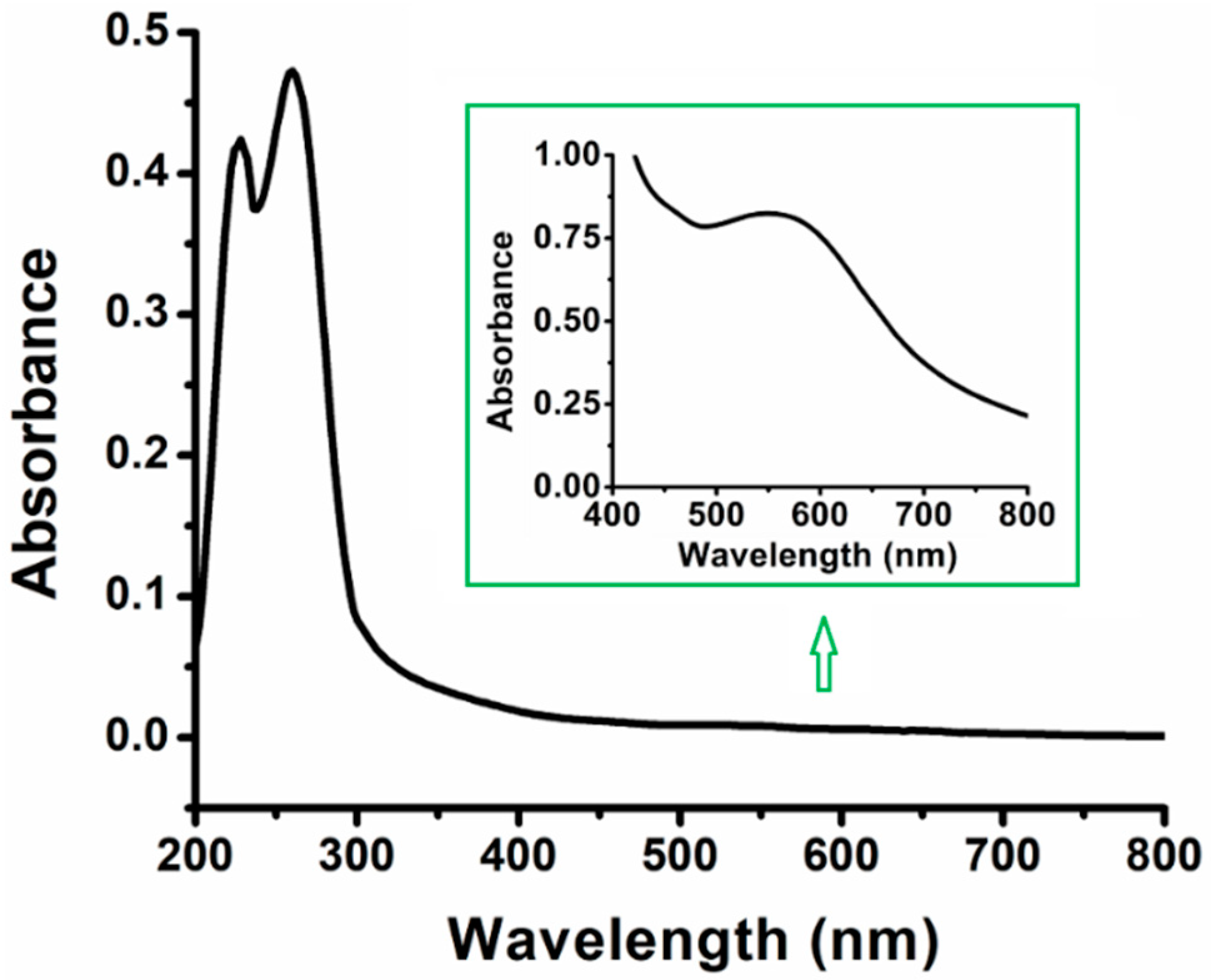
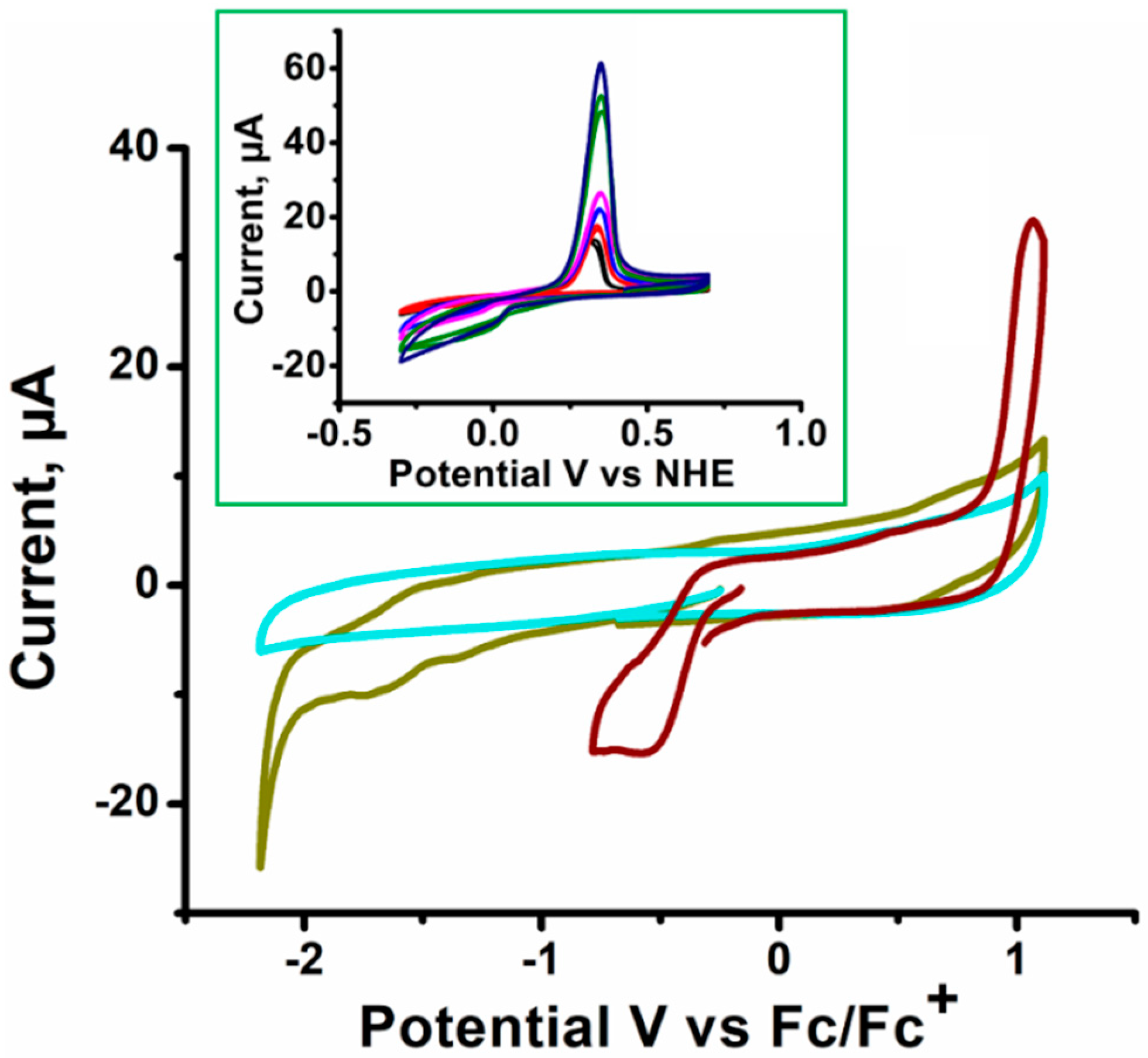
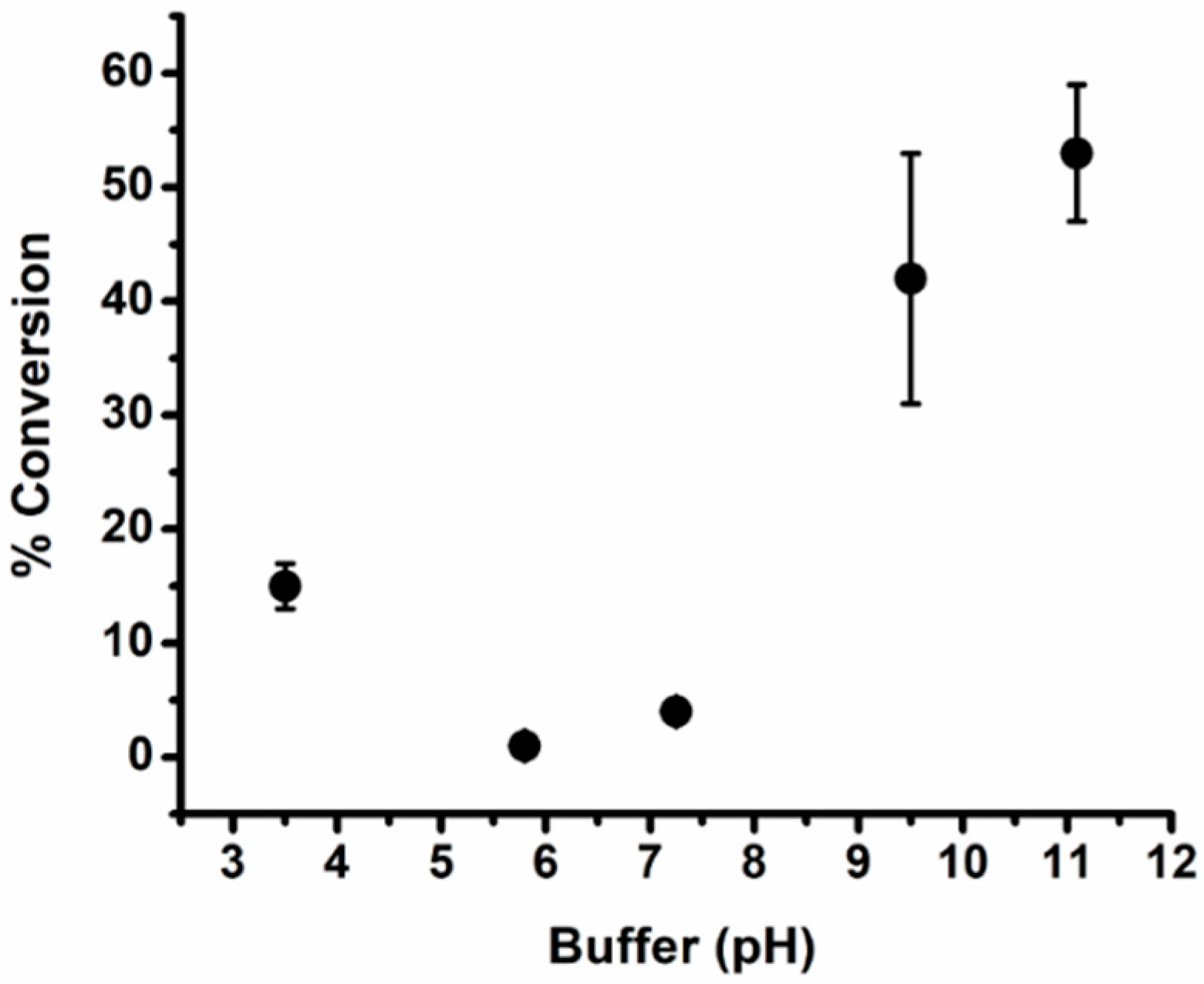
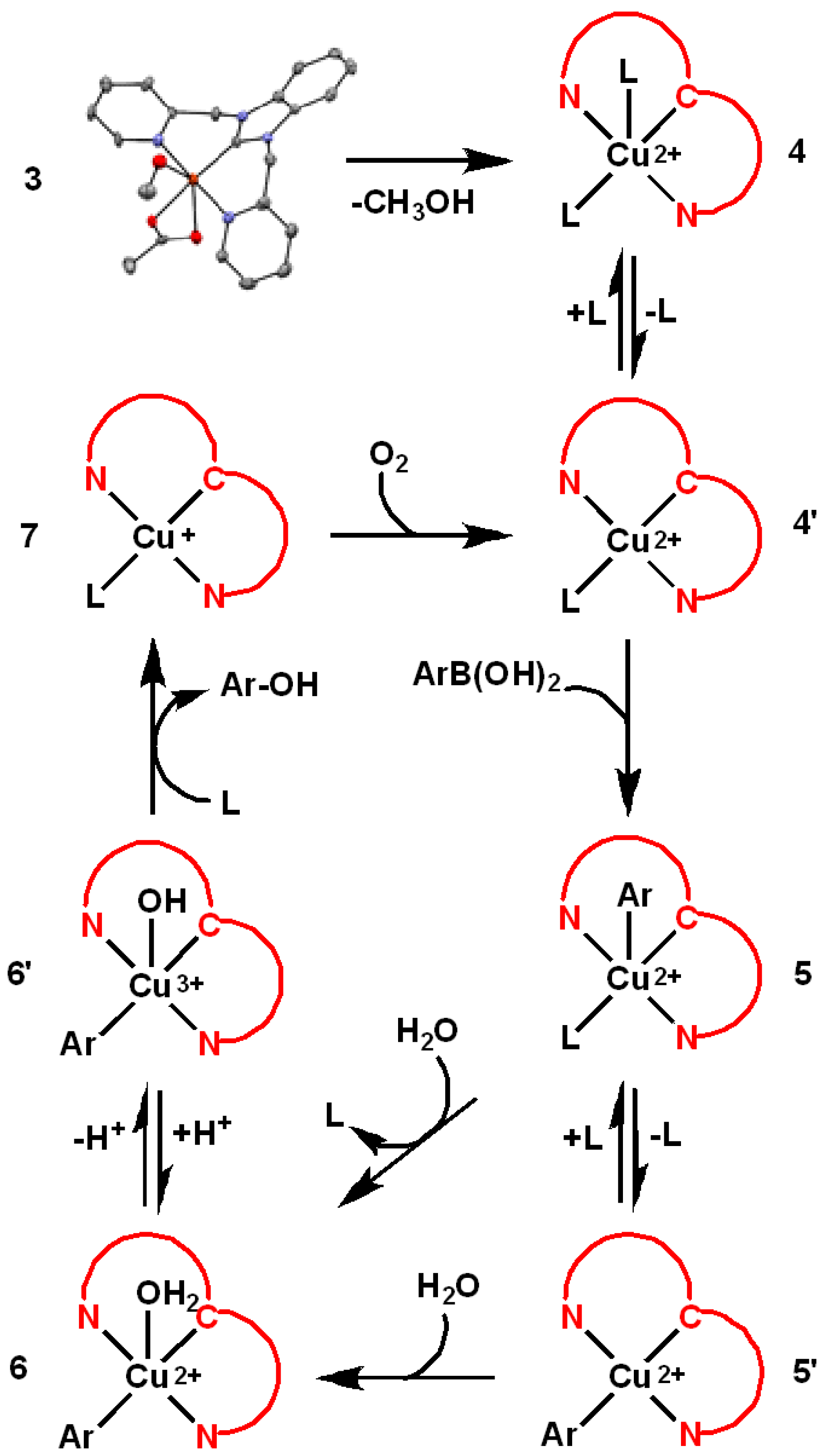
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, B.; Zhang, Y.; Xu, D.; Chen, W. Facile Synthesis of Metal N-Heterocyclic Carbene Complexes. Chem. Commun. 2011, 47, 2883–2885. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.E.; Jahnke, M.C. Heterocyclic carbenes: Synthesis and coordination chemistry. Angew. Chem. Int. Ed. 2008, 47, 3122–3172. [Google Scholar] [CrossRef]
- Hanh, F.E.; Jahnke, M.C. Heterocyclische Carbene—Synthese und Koordinationschemie. Angew. Chem. 2008, 120, 3166–3216. [Google Scholar]
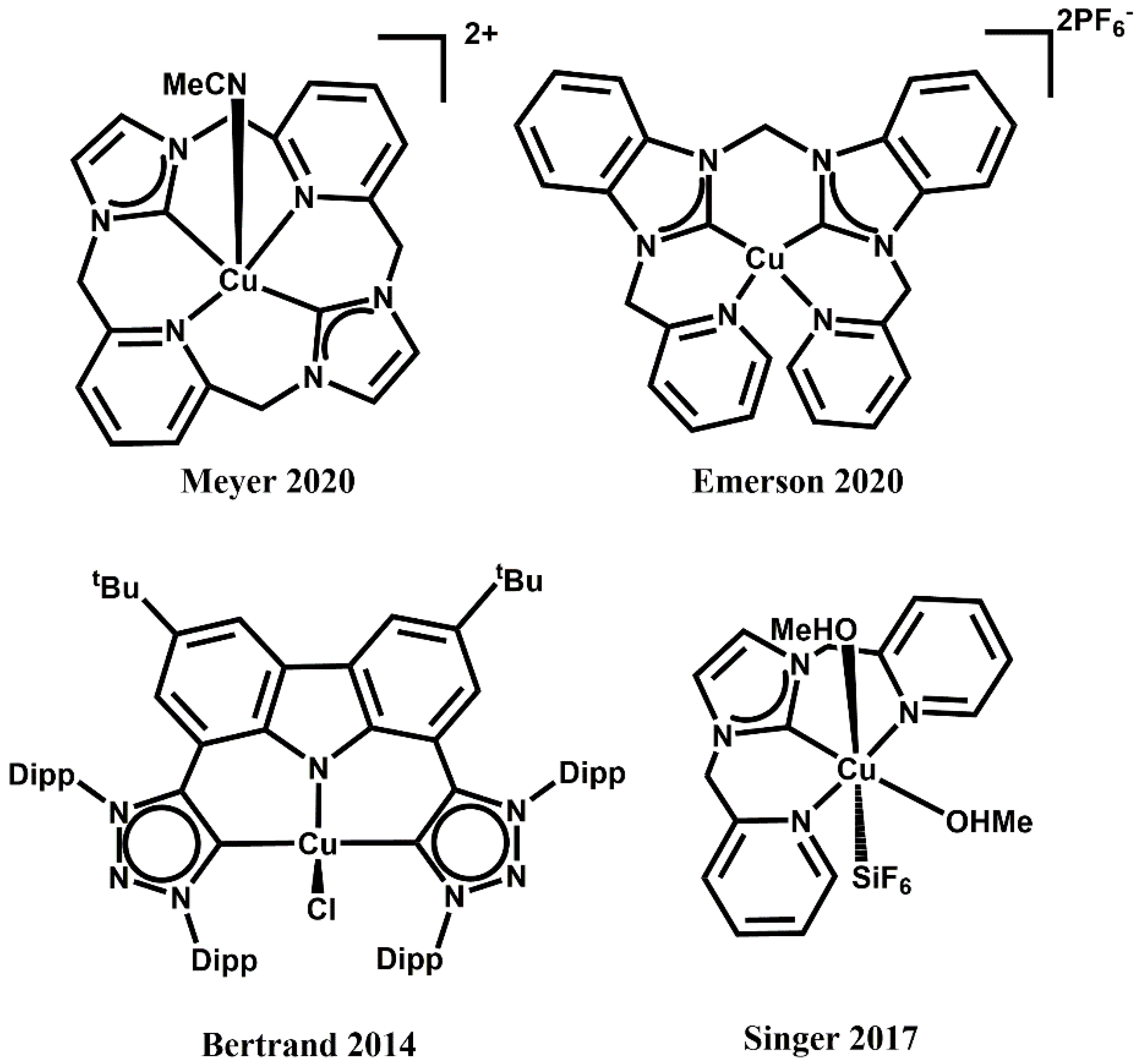
- Cope, J.D.; Sheridan, P.E.; Galloway, C.J.; Awoyemi, R.F.; Stokes, S.L.; Emerson, J.P. Synthesis and Characterization of a Tetradentate, N-Heterocyclic Carbene Copper(II) Complex and Its Use as a Chan–Evans–Lam Coupling Catalyst. Organometallics 2020, 39, 4457–4464. [Google Scholar] [CrossRef]
- Monnier, F.; Taillefer, M. Catalytic C–C, C–N, and C–O Ullmann-Type Coupling Reactions. Angew. Chem. Int. Ed. 2009, 48, 6954–6971. [Google Scholar] [CrossRef]
- Monnier, F.; Taillefer, M. Katalytische C–C–, C–N– und C–O-Ullmann-Kupplungen. Angew. Chem. 2009, 121, 7088–7105. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Dias, H.R.; Calabrese, J.C.; Davidson, F. Homoleptic carbene-silver (I) and carbene-copper (I) complexes. Organometallics 1993, 12, 3405–3409. [Google Scholar] [CrossRef]
- Lin, J.C.; Huang, R.T.; Lee, C.S.; Bhattacharyya, A.; Hwang, W.S.; Lin, I.J. Coinage metal–N-heterocyclic carbene complexes. Chem. Rev. 2009, 109, 3561–3598. [Google Scholar] [CrossRef]
- Liu, B.; Ma, X.; Wu, F.; Chen, W. Simple synthesis of neutral and cationic Cu-NHC complexes. Dalton Trans. 2015, 44, 1836–1844. [Google Scholar] [CrossRef]
- Lazreg, F.; Nahra, F.; Cazin, C.S. Copper–NHC complexes in catalysis. Coord Chem. Rev. 2015, 293, 48–79. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, Z.S.; Anneser, M.R.; Kaiser, F.; Altmann, P.J.; Hofmann, B.J.; Schlagintweit, J.F.; Grivani, G.; Kühn, F.E. A bench stable formal Cu (iii) N-heterocyclic carbene accessible from simple copper(II) acetate. Chem. Sci. 2018, 9, 8307–8314. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Castro-Rodriguez, I.; Meyer, K. Copper complexes of nitrogen-anchored tripodal N-heterocyclic carbene ligands. J. Am. Chem. Soc. 2003, 125, 12237–12245. [Google Scholar] [CrossRef]
- Liu, Y.; Resch, S.G.; Klawitter, I.; Cutsail, G.E., III; Demeshko, S.; Dechert, S.; Kühn, F.E.; DeBeer, S.; Meyer, F. An Adaptable N-Heterocyclic Carbene Macrocycle Hosting Copper in Three Oxidation States. Angew. Chem. Int. Ed. 2020, 132, 5696–5705. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Resch, S.G.; Klawitter, I.; Cutsail, G.E., III; Demeshko, S.; Dechert, S.; Kühn, F.E.; DeBeer, S.; Meyer, F. An Adaptable N-Heterocyclic Carbene Macrocycle Hosting Copper in Three Oxidation States. Angew. Chem. 2020, 132, 5745–5754. [Google Scholar] [CrossRef] [Green Version]
- Bezuidenhout, D.I.; Kleinhans, G.; Guisado-Barrios, G.; Liles, D.C.; Ung, G.; Bertrand, G. Isolation of a potassium bis (1, 2, 3-triazol-5-ylidene) carbazolide: A stabilizing pincer ligand for reactive late transition metal complexes. Chem. Comm. 2014, 50, 2431–2433. [Google Scholar] [CrossRef] [Green Version]
- O’Hearn, D.J.; Singer, R.D. Direct synthesis of a copper(II) N-heterocyclic carbene complex in air. Organometallics 2017, 36, 3175–3177. [Google Scholar] [CrossRef]
- Zhu, C.; Falck, J.R. Transition Metal-Free ipso-Functionalization of Arylboronic Acids and Derivatives. Adv. Synth. Catal. 2014, 356, 2395–2410. [Google Scholar] [CrossRef] [Green Version]
- Vantourout, J.C.; Miras, H.N.; Isidro-Llobet, A.; Sproules, S.; Watson, A.J. Spectroscopic studies of the Chan–Lam amination: A mechanism-inspired solution to boronic ester reactivity. J. Am. Chem. Soc. 2017, 139, 4769–4779. [Google Scholar] [CrossRef] [Green Version]
- West, M.J.; Fyfe, J.W.; Vantourout, J.C.; Watson, A.J. Mechanistic development and recent applications of the Chan–Lam amination. Chem. Rev. 2019, 119, 12491–12523. [Google Scholar] [CrossRef]
- Akatyev, N.; Il’in, M.; Il’in, M.; Peregudova, S.; Peregudov, A.; Buyanovskaya, A.; Kudryavtsev, K.; Dubovik, A.; Grinberg, V.; Orlov, V.; et al. Chan-Evans-Lam C–N Coupling Promoted by a Dinuclear Positively Charged Cu (II) Complex. Catalytic Performance and Some Evidence for the Mechanism of CEL Reaction Obviating Cu (III)/Cu (I) Catalytic Cycle. ChemCatChem 2020, 12, 3010–3021. [Google Scholar] [CrossRef]
- Vummaleti, S.V.; Nelson, D.J.; Poater, A.; Gómez-Suárez, A.; Cordes, D.B.; Slawin, A.M.; Nolan, S.P.; Cavallo, L. What can NMR spectroscopy of selenoureas and phosphinidenes teach us about the π-accepting abilities of N-heterocyclic carbenes? Chem. Sci. 2015, 6, 1895–1904. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic carbene complexes in C–H activation reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar] [CrossRef]
- Karthick, K.; Nithiyanantham, U.; Ede, S.R.; Kundu, S. DNA aided formation of aggregated Nb2O5 nanoassemblies as anode material for dye sensitized solar cell (DSSC) and supercapacitor applications. ACS Sustain. Chem. Eng. 2016, 4, 3174–3188. [Google Scholar] [CrossRef]
- Sharma, M.; Saikia, G.; Ahmed, K.; Gogoi, S.R.; Puranik, V.G.; Islam, N.S. Vanadium-based polyoxometalate complex as a new and efficient catalyst for phenol hydroxylation under mild conditions. New J. Chem. 2018, 42, 5142–5152. [Google Scholar] [CrossRef]
- Dandia, A.; Sharma, R.; Saini, P.; Badgoti, R.S.; Rathore, K.S.; Parewa, V. The graphite-catalyzed ipso-functionalization of arylboronic acids in an aqueous medium: Metal-free access to phenols, anilines, nitroarenes, and haloarenes. RSC Adv. 2021, 11, 18040–18049. [Google Scholar] [CrossRef]
- Anderson, K.W.; Ikawa, T.; Tundel, R.E.; Buchwald, S.L. The selective reaction of aryl halides with KOH: Synthesis of phenols, aromatic ethers, and benzofurans. J. Am. Chem. Soc. 2006, 128, 10694–10695. [Google Scholar] [CrossRef]
- Adhikary, S.D.; Samanta, T.; Roymahapatra, G.; Loiseau, F.; Jouvenot, D.; Giri, S.; Chattaraj, P.K.; Dinda, J. Synthesis, structure and electrochemical behaviour of Ru (ii)-and Pt (ii)-carbene complexes of the NCN-pincer 1, 3-bis (2-pyridylmethyl)-1 H-benzimidazolium chloride. New J. Chem. 2010, 34, 1974–1980. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Palaniandavar, M. Interaction of rac-[Cu (diimine) 3] 2+ and rac-[Zn (diimine) 3] 2+ complexes with CT DNA: Effect of fluxional Cu (II) geometry on DNA binding, ligand–promoted exciton coupling and prominent DNA cleavage. Dalton Trans. 2008, 29, 3866–3878. [Google Scholar] [CrossRef]
- Sharma, M.; Ganeshpandian, M.; Majumder, M.; Tamilarasan, A.; Sharma, M.; Mukhopadhyay, R.; Islam, N.S.; Palaniandavar, M. Octahedral copper (ii)-diimine complexes of triethylenetetramine: Effect of stereochemical fluxionality and ligand hydrophobicity on Cu II/Cu I redox, DNA binding and cleavage, cytotoxicity and apoptosis-inducing ability. Dalton Trans. 2020, 49, 8282–8297. [Google Scholar] [CrossRef]
- Murphy, B.; Aljabri, M.; Ahmed, A.M.; Murphy, G.; Hathaway, B.J.; Light, M.E.; Geilbrich, T.; Hursthouse, M.B. Structural systematics of the [Cu (chelate) 3][Y] 2 series. An interesting crystallographic structural insight involving vibronic coupling and the Jahn–Teller effect (JTE). The syntheses and low temperature crystal structures of tris (2, 2′ bipyridyl) copper (II) tetraphenylborate and tris (2, 2′ bipyridyl) zinc (II) tetraphenylborate. Dalton Trans. 2006, 2, 357–367. [Google Scholar]
- Sharma, M.; Ganeshpandian, M.; Sanjeev, A.; Tamilarasan, A.; Mattaparthi, V.S.K.; Islam, N.S.; Palaniandavar, M. Bis-and mixed-ligand copper (II) complexes of nalidixic acid the antibacterial drug: Mode of nalidixate coordination determines DNA binding and cleavage and cytotoxicity. Inorg. Chim. Acta 2020, 504, 119450. [Google Scholar] [CrossRef]
- Brudenell, S.J.; Spiccia, L.; Bond, A.M.; Comba, P.; Hockless, D.C. Structural, EPR, and electrochemical studies of binuclear copper (II) complexes of bis (pentadentate) ligands derived from bis (1, 4, 7-triazacyclonane) macrocycles. Inorg. Chem. 1998, 37, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Tas, E.; Aslanoglu, M.; Kilic, A.; Kara, Z. Synthesis, spectroscopic and electrochemical studies of copper (II) and cobalt (II) complexes of three unsymmetrical vic-dioximes ligands. J. Coord. Chem. 2006, 59, 861–872. [Google Scholar] [CrossRef]
- Petković, B.; Stevanović, S.; Budimir, M.; Sovilj, S.P.; Jovanović, V.M. Electrochemical examination of copper (II) complexes with octaazamacrocyclic ligand and heterocyclic dithiocarbamate. Electroanalysis 2012, 24, 1605–1612. [Google Scholar] [CrossRef]
- Santos, M.A.; Gaspar, M.; Gonçalves, M.L. Electrochemistry of copper (II) complexes of dioxocyclam and dihydroxamate derivative. Electroanal. Int. J. Devoted Fundam. Pract. Asp. Electroanal. 2000, 12, 66–71. [Google Scholar] [CrossRef]
- Smart, R.B.; Weber, J.H. Differential pulse anodic stripping voltammetry of copper (II) at the glassy carbon electrode. Anal. Chim. Acta 1980, 115, 331–336. [Google Scholar] [CrossRef]
- Štulikova, M. The deposition and stripping of mercury on a glassy carbon rotating disk electrode. J. Electroanal. Chem. Interfacial Electrochem. 1973, 48, 33–45. [Google Scholar] [CrossRef]
- Atli, D.D. Synthesis, characterization and catalytic properties of cationic N-heterocyclic carbene silver complexes. Turk. J. Chem. 2021, 45, 577–584. [Google Scholar] [CrossRef]
- Cheng, J.; Wang, L.; Wang, P.; Deng, L. High-oxidation-state 3d metal (Ti–Cu) complexes with N-heterocyclic carbene ligation. Chem. Rev. 2018, 118, 9930–9987. [Google Scholar] [CrossRef]
- Smith, J.M.; Long, J.R. First-row transition metal complexes of the strongly donating pentadentate ligand PY4Im. Inorg. Chem. 2010, 49, 11223–11230. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Chandra Shit, S.; Mandal, H.; Rabeah, J.; Kashyap, S.S.; Nailwal, Y.; Shinde, D.B.; Lai, Z.; Mondal, J. Benzothiazole-Linked Metal-Free Covalent Organic Framework Nanostructures for Visible-Light-Driven Photocatalytic Conversion of Phenylboronic Acids to Phenols. ACS Appl. Nano Mater. 2021, 4, 11732–11742. [Google Scholar] [CrossRef]
- Feizi Mohazzab, B.; Jaleh, B.; Nasrollahzadeh, M.; Issaabadi, Z. Journey on greener pathways via synthesis of Pd/KB polymeric nanocomposite as a recoverable catalyst for the ligand-free oxidative hydroxylation of phenylboronic acid and Suzuki–Miyaura coupling reaction in green solvents. Catal. Lett. 2019, 149, 169–179. [Google Scholar] [CrossRef]
- Upadhyay, R.; Singh, D.; Maurya, S.K. Highly efficient heterogeneous V2O5@ TiO2 catalyzed the rapid transformation of boronic acids to phenols. Eur. J. Org. Chem. 2021, 2021, 3925–3931. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, J.; Liu, Y.; Lin, L.; Lei, L.; Zhang, H.; Wang, Z.; Cheng, H.; Wang, P.; Zheng, Z.; et al. Enhanced photocatalytic driven hydroxylation of phenylboric acid to phenol over pyrenetetrasulfonic acid intercalated ZnAl-LDHs. J. Colloid Interface Sci. 2022, 610, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Sadhasivam, V.; Harikrishnan, M.; Elamathi, G.; Balasaravanan, R.; Murugesan, S.; Siva, A. Copper nanoparticles supported on highly nitrogen-rich covalent organic polymers as heterogeneous catalysts for the ipso-hydroxylation of phenyl boronic acid to phenol. New J. Chem. 2020, 44, 6222–6231. [Google Scholar] [CrossRef]
- Shin, E.J.; Joo, S.R.; Kim, S.H. Cooperation of biopolymer chitosan with hydrogen peroxide for ipso-hydroxylation of arylboronic acids under green conditions. Tetrahedron Lett. 2019, 60, 1509–1513. [Google Scholar] [CrossRef]









 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst Loading (Mol%) | Time (h) | Temperature (°C) | % Conversion | TON b | Base (Equivalents) |
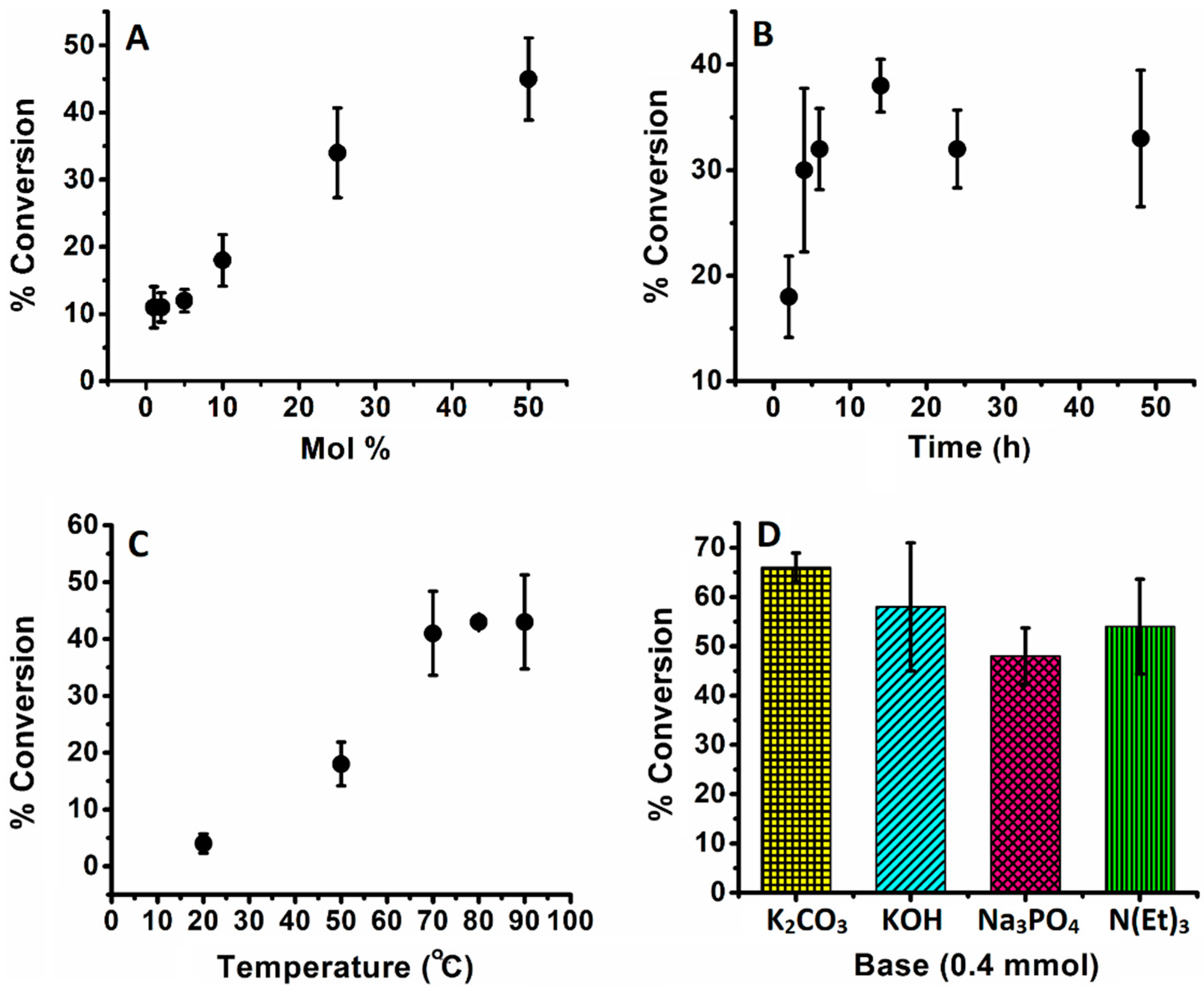
| 1 | 1 | 2 | 50 | 11 | 11 | - |
| 2 | 2 | 2 | 50 | 11 | 5.5 | - |
| 3 | 5 | 2 | 50 | 12 | 2.4 | - |
| 4 | 10 | 2 | 50 | 18 | 1.8 | - |
| 5 | 25 | 2 | 50 | 34 | 1.4 | - |
| 6 | 50 | 2 | 50 | 45 | 0.9 | - |
| 7 | 10 | 4 | 50 | 30 | 3 | - |
| 8 | 10 | 6 | 50 | 32 | 3.2 | - |
| 9 | 10 | 14 | 50 | 38 | 3.8 | - |
| 10 | 10 | 24 | 50 | 32 | 3.2 | - |
| 11 | 10 | 48 | 50 | 33 | 3.3 | - |
| 12 | 10 | 6 | RT | 4 | 0.4 | - |
| 13 | 10 | 6 | 70 | 41 | 4.1 | - |
| 14 | 10 | 6 | 80 | 43 | 4.3 | - |
| 15 | 10 | 6 | 90 | 43 | 4.3 | - |
| 16 | 10 | 6 | 70 | 53 | 5.3 | K2CO3 (1) |
| 17 | 10 | 6 | 70 | 66 | 6.6 | K2CO3 (4) |
| 18 | 10 | 6 | 70 | 58 | 5.8 | KOH (4) |
| 19 | 10 | 6 | 70 | 48 | 4.8 | Na3PO4 (4) |
| 20 | 10 | 6 | 70 | 54 | 5.4 | NEt3 (4) |
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Catalyst Loading (mol %) | % Conversion | Base (Equivalents) |
| 1 | CuCl2 | 10 | 31 | - |
| 2 | CuCl2 + Bpy (1:1) | 10 | 36 | - |
| 3 | CuCl2 + Bpy (1:2) | 10 | 32 | - |
| 4 | CuCl2 + Phen (1:1) | 10 | 34 | - |
| 5 | CuCl2 + Phen (1:2) | 10 | 37 | - |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, M.; Adhikari, B.; Awoyemi, R.F.; Perkins, A.M.; Duckworth, A.K.; Donnadieu, B.; Wipf, D.O.; Stokes, S.L.; Emerson, J.P. Copper(II) NHC Catalyst for the Formation of Phenol from Arylboronic Acid. Chemistry 2022, 4, 560-575. https://doi.org/10.3390/chemistry4020040
Sharma M, Adhikari B, Awoyemi RF, Perkins AM, Duckworth AK, Donnadieu B, Wipf DO, Stokes SL, Emerson JP. Copper(II) NHC Catalyst for the Formation of Phenol from Arylboronic Acid. Chemistry. 2022; 4(2):560-575. https://doi.org/10.3390/chemistry4020040
Chicago/Turabian StyleSharma, Mitu, Bhupendra Adhikari, Raymond Femi Awoyemi, Amanda M. Perkins, Alison K. Duckworth, Bruno Donnadieu, David O. Wipf, Sean L. Stokes, and Joseph P. Emerson. 2022. "Copper(II) NHC Catalyst for the Formation of Phenol from Arylboronic Acid" Chemistry 4, no. 2: 560-575. https://doi.org/10.3390/chemistry4020040