
Structures and Bonding in Hexacarbonyl Diiron Polyenes: Cycloheptatriene and 1,3,5-Cyclooctatriene


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
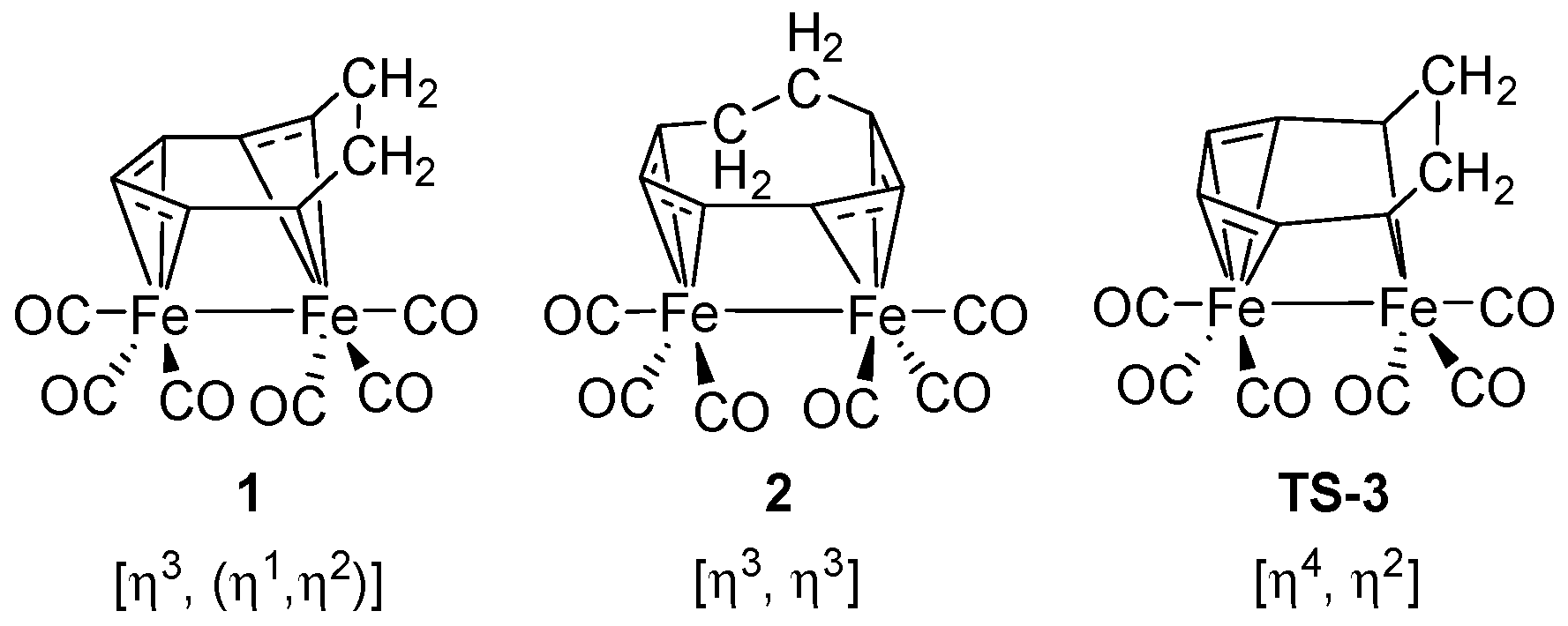{kind=link}
1. Introduction
2. Computational Methods
3. Results and Discussion
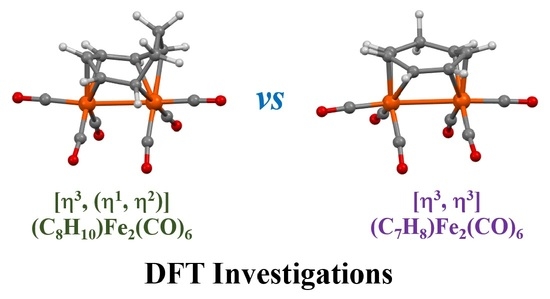
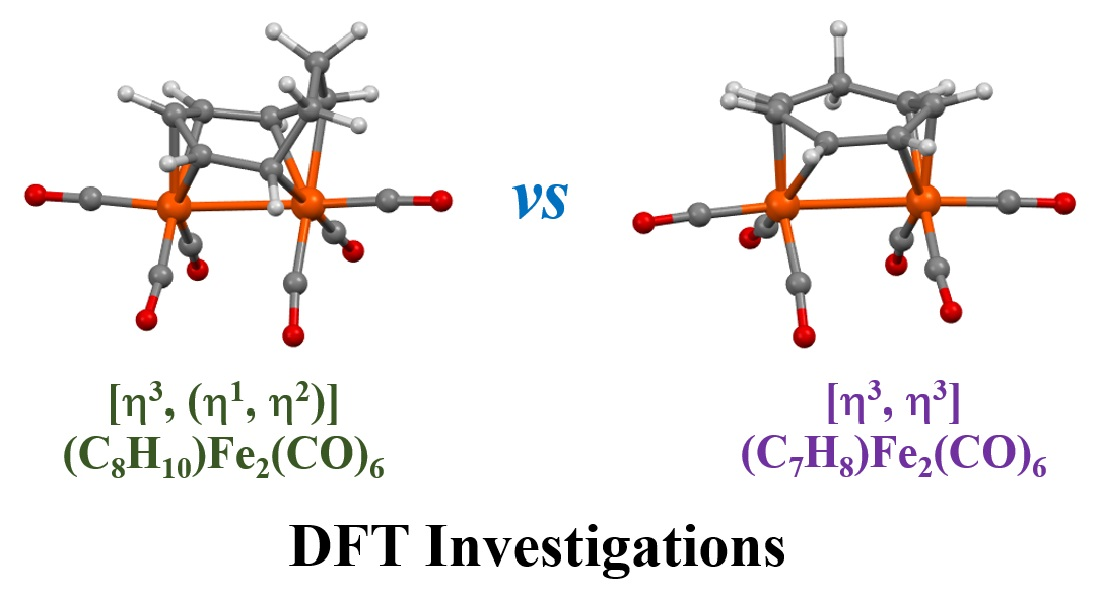
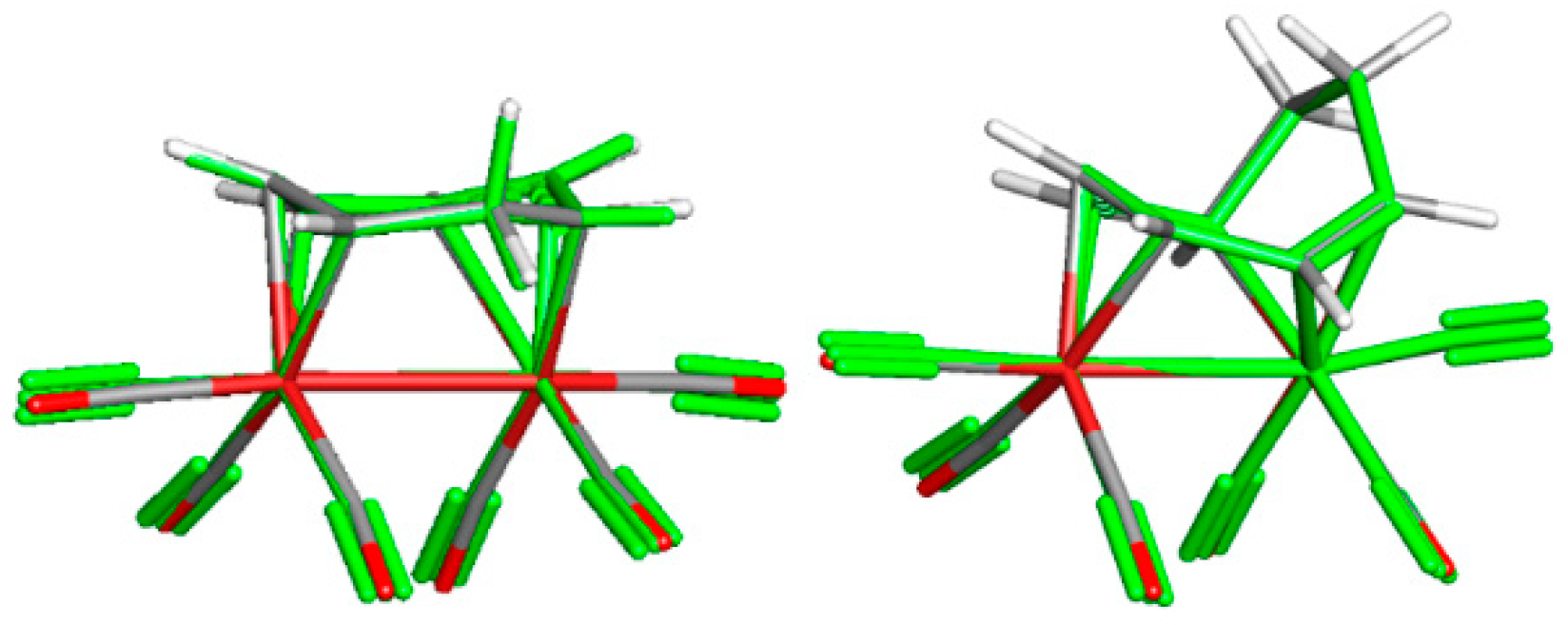
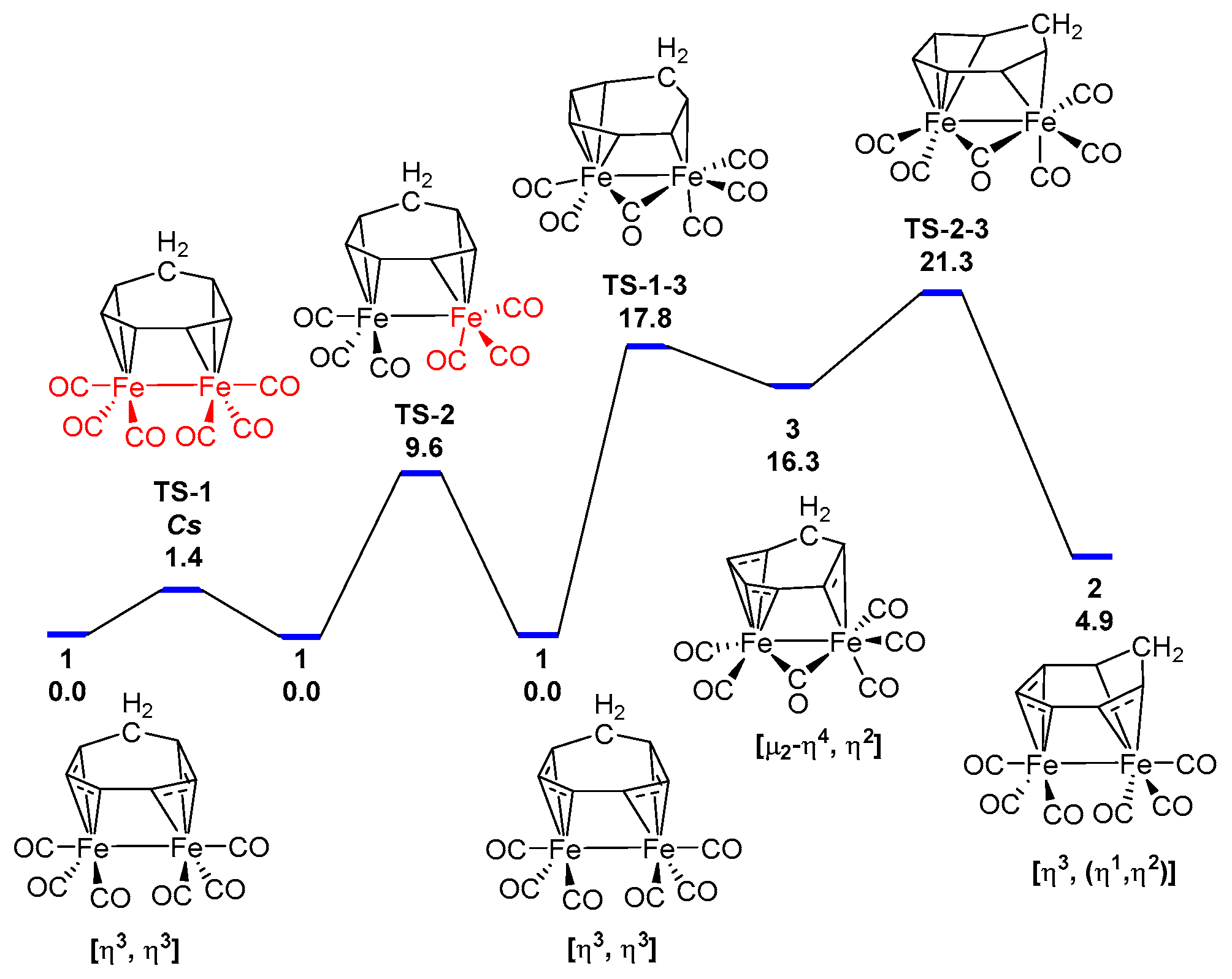
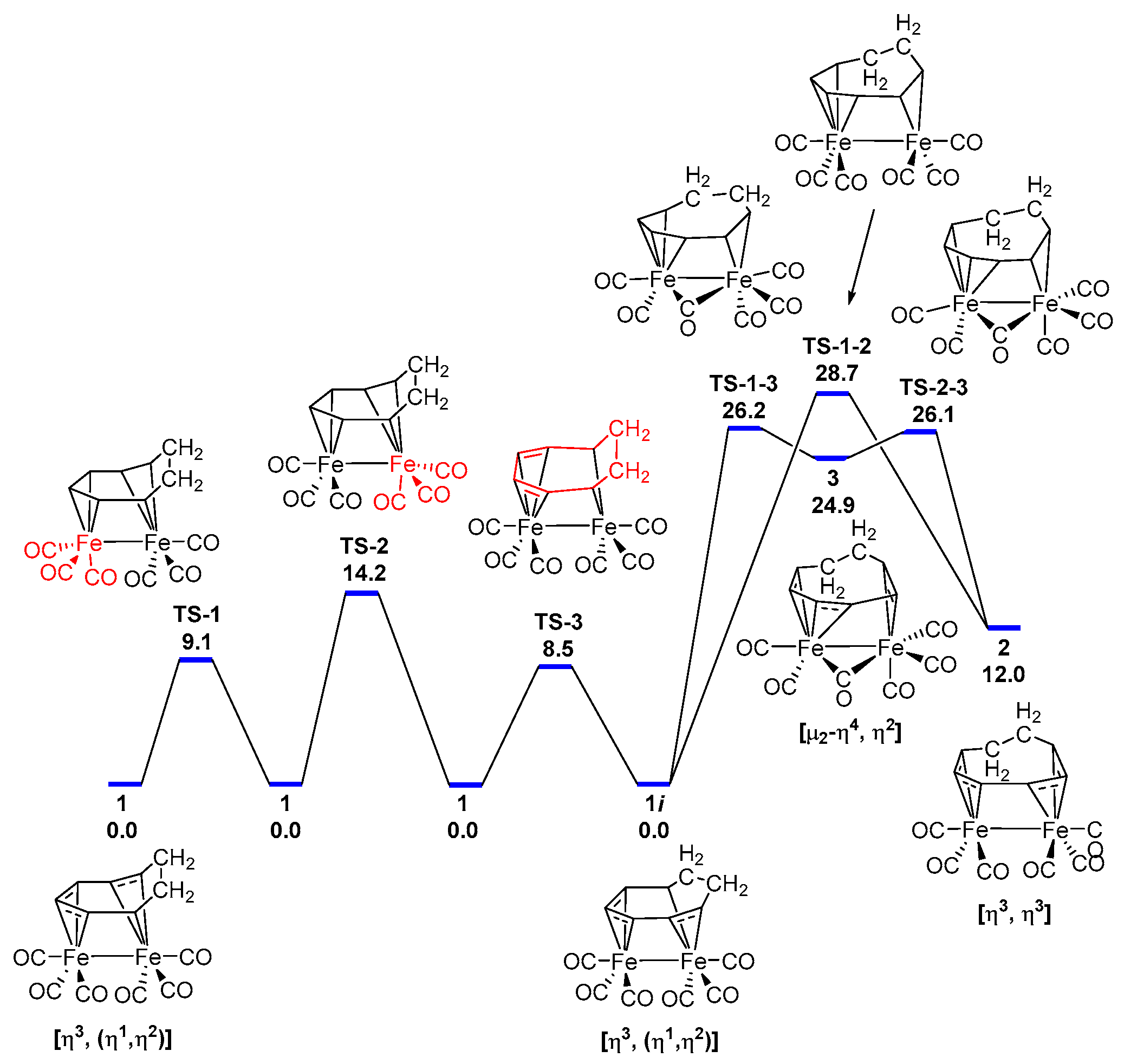
3.1. The Interconversions of (C7H8)Fe2(CO)6
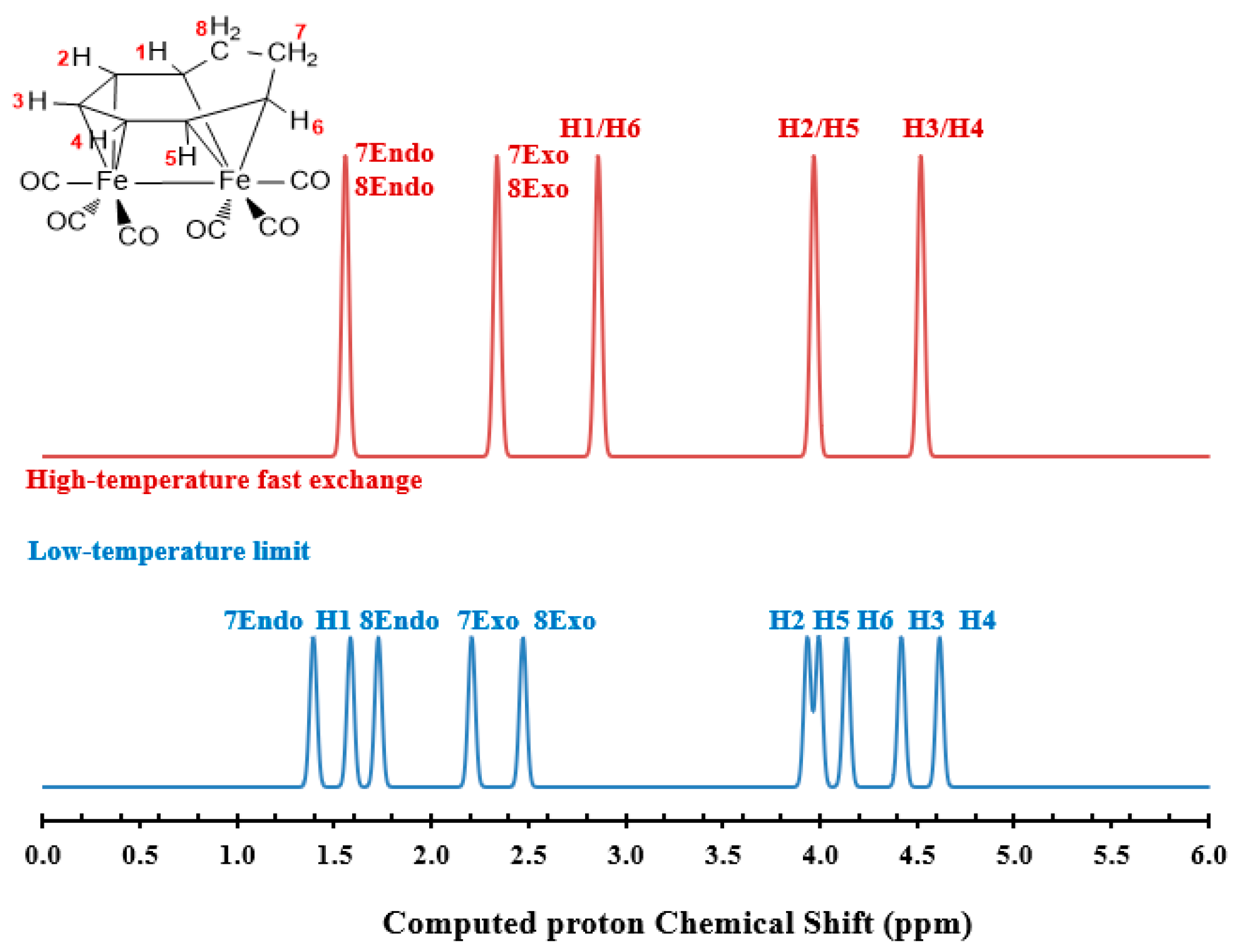
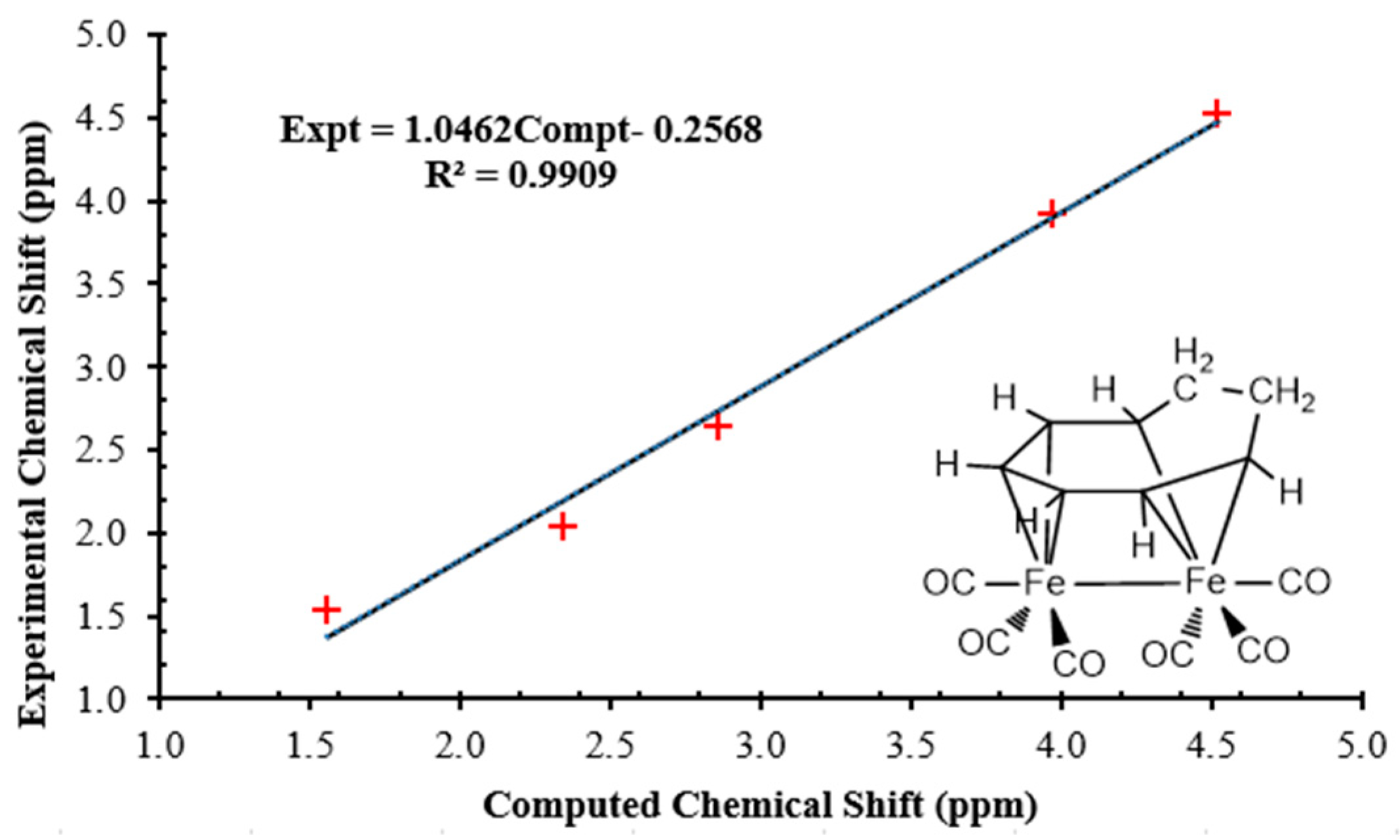
3.2. Dynamic Fluxionality of (C8H10)Fe2(CO)6
3.3. Interpretations of the Dynamic Fluxionality
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, R.B. Organometallic Chemistry of the Transition Metals. V. New Iron Carbonyl Complexes of Cycloöctatriene Derivatives. Inorg. Chem. 1963, 2, 807–810. [Google Scholar] [CrossRef]
- Emerson, G.F.; Mahler, J.E.; Pettit, R.; Collins, R. Organometallic Complexes of the Type Triene-Fe2(CO)6. J. Am. Chem. Soc. 1964, 86, 3590–3591. [Google Scholar] [CrossRef]
- Cotton, F.A.; Edwards, W.T. Crystal and molecular structure of (1,3,5-cyclooctatriene)diiron hexacarbonyl. J. Am. Chem. Soc. 1969, 91, 843–847. [Google Scholar] [CrossRef]
- Cotton, F.A.; Marks, T.J. Stereochemically nonrigid organometallic molecules XXV. The low-temperature PMR spectrum of cis-(1,2,6-trihapto:3,4,5-trihapto-1,3,5-cyclooctatriene)hexacarbonyldiiron. J. Organomet. Chem. 1969, 19, 237–240. [Google Scholar] [CrossRef]
- Cotton, F.A. A Half-Century of Nonclassical Organometallic Chemistry: A personal Perspective. Inorg. Chem. 2002, 41, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Cotton, F.A.; DeBoer, B.G.; Marks, T.J. Stereochemically nonrigid organometallic molecules. XXIX. Cycloheptatrienediiron hexacarbonyl. J. Am. Chem. Soc. 1971, 93, 5069–5075. [Google Scholar] [CrossRef]
- Komiya, S.; Planas, J.G.; Onuki, K.; Lu, Z.; Hirano, M. Versatile Coordination Modes and Transformations of the Cyclooctatriene Ligand in Ru(C8H10)L3 (L = Tertiary Phosphine). Organometallics 2000, 19, 4051–4059. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Xu, Q.; Sun, J.; Chen, J. Cyclooctatetraene (COT)-Coordinated Diiron Carbene Complexes and Their Remarkable Thermolysis Reactions. Organometallics 2005, 24, 933–944. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Dunlap, B.I. Fitting the Coulomb potential variationally in Xα molecular calculations. J. Chem. Phys. 1983, 78, 3140–3142. [Google Scholar] [CrossRef]
- Dunlap, B.I. Robust and variational fitting: Removing the four-center integrals from center stage in quantum chemistry. J. Mol. Struct. THEOCHEM 2000, 529, 37–40. [Google Scholar] [CrossRef]
- Couty, M.; Hall, M.B. Basis sets for transition metals: Optimized outer p functions. J. Comput. Chem. 1996, 17, 1359–1370. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- The 6-31G(d′) Basis Set Has the Exponent of D Polarization Functions for C, N, O, and F Taken from the 6-311G(d) Basis Sets, Instead of the Original Arbitrarily Assigned Exponent of 0.8 Used in the 6-31G(d) Basis Sets. For H, the 6-31G(d′) Keyword Utilizes the 6-31G(d) Basis Sets.
- Liang, G.; Webster, C.E. The Missing Agostomer in the Fluxionality of Cyclohexenyl Manganese Tricarbonyl. J. Organomet. Chem. 2018, 864, 128–135. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L. Theoretical study of Re(IV) and Ru(II) bis-isocyanide complexes and their reactivity in cycloaddition reactions with nitrones. Inorg. Chim. Acta 2012, 380, 78–89. [Google Scholar] [CrossRef]
- Novikov, A.S.; Kuznetsov, M.L.; Rocha, B.G.M.; Pombeiro, A.J.L.; Shul’pin, G.B. Oxidation of olefins with H2O2 catalysed by salts of group III metals (Ga, In, Sc, Y and La): Epoxidation versus hydroperoxidation. Catal. Sci. Technol. 2016, 6, 1343–1356. [Google Scholar] [CrossRef]
- Liang, G.; Hollis, T.K.; Webster, C.E. Computational Analysis of the Intramolecular Oxidative Amination of an Alkene Catalyzed by the Extreme π-loading N-Heterocyclic Carbene Pincer Tantalum(V) Bis(imido) Complex. Organometallics 2018, 37, 1671–1681. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. 1. Gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient Implementation of the Gauge-Independent Atomic Orbital Method for NMR Chemical Shift Calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Roy, L.E.; Hay, P.J.; Martin, R.L. Revised basis sets for the LANL effective core potentials. J. Chem. Theory Comput. 2008, 4, 1029–1031. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-Consistent Molecular-Orbital Methods. 25. Supplementary Functions for Gaussian-Basis Sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. Ruthenium Complexes of Stibino Derivatives of Carboxylic Amides: Synthesis and Characterization of Bidentate Sb,E, Tridentate Sb,E2, and Tetradentate Sb,E3 (E = N and O) Ligands and Their Reactivity Toward [RuCl2(PPh3)3]. Inorg. Chem. 2020, 59, 6359–6375. [Google Scholar] [CrossRef]
- Škríba, A.; Jašík, J.; Andris, E.; Roithová, J. Interaction of Ruthenium(II) with Terminal Alkynes: Benchmarking DFT Methods with Spectroscopic Data. Organometallics 2016, 35, 990–994. [Google Scholar] [CrossRef]
- Cotton, F.A.; Hunter, D.L. Carbon-13 nuclear magnetic resonance study of the fluxional character of the .eta.6-(bicyclo[6.2.0]dodeca-2,4,6-triene)hexacarbonyldiiron(Fe-Fe) and a triethylphosphine derivative thereof, and the crystal structure of the latter. J. Am. Chem. Soc. 1975, 97, 5739–5746. [Google Scholar] [CrossRef]
- Deganello, G.; Lewis, J.; Parker, D.G.; Sandrini, P.L. The fluxional behaviour of η6(bicyclo[6.2.0]deca-2,4,6-triene) hexacarbonyl diruthenium (Ru-Ru) by 1H and 13C-NMR techniques. Inorg. Chem. Acta 1977, 24, 165–171. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Liang, G. Structures and Bonding in Hexacarbonyl Diiron Polyenes: Cycloheptatriene and 1,3,5-Cyclooctatriene. Chemistry 2022, 4, 447-453. https://doi.org/10.3390/chemistry4020033
Zhang M, Liang G. Structures and Bonding in Hexacarbonyl Diiron Polyenes: Cycloheptatriene and 1,3,5-Cyclooctatriene. Chemistry. 2022; 4(2):447-453. https://doi.org/10.3390/chemistry4020033
Chicago/Turabian StyleZhang, Min, and Guangchao Liang. 2022. "Structures and Bonding in Hexacarbonyl Diiron Polyenes: Cycloheptatriene and 1,3,5-Cyclooctatriene" Chemistry 4, no. 2: 447-453. https://doi.org/10.3390/chemistry4020033