1. Introduction
Free radicals are ubiquitous in our environment, and our body is exposed to them daily. Oxygen-centered radicals cause oxidative stress and are produced due to endogenous sources as a result of metabolism as well as exogenous sources, such as exposure to smoke and radiation. Thus, we are continuously exposed to oxidative stress (OS). To combat this OS, organisms have a defense system mediated by antioxidant enzymes and molecules. However, some of the free radicals escape from these antioxidant defense systems, due to which we are exposed to a chronic low dose of oxidants, which is the etiology of cancer [
1,
2]. Nutraceuticals are available from different foods, fruits, and vegetables and have been shown to possess an antioxidant capacity. Thus, supplementation with these nutraceuticals can enhance the capacity to fight against oxidative stress [
3,
4]. Myricetin falls under the group of flavonols and is found in food items such as vegetables, fruits, and tea and has shown potent activity against many radicals, even at low concentrations, compared to other flavonoids [
5,
6]. The myricetin structure has two aromatic rings, A and B, that are combined by a three-carbon chain forming a cyclic ring C, and reports suggest that the presence of more hydroxyl groups is one of the reasons for myricetin to be a potent antioxidant [
7]. The drawback of myricetin being used for oral consumption is that it is poorly soluble in water and its bioavailability is low. Other than protective effects, myricetin also shows pro-oxidant activity, which is harmful to the cells as well as biomolecules [
8].
Nanotechnology is a field of science that is emerging rapidly, yielding enormous applications in the biomedical field for the treatment of many diseases, such as Alzheimer’s disease and Parkinson’s disease [
9,
10]. The different physical parameters, such as pH and temperature, can be altered to tune the size and surface properties of these nanostructures [
11,
12]. Different nanostructures include liposomes, micelles, nanoparticles, nanosheets, nanorods, solid lipid nanoparticles, niosomes, vesicles, microporous silica, magnetic nanoparticles, metal, and metal-oxide-based nanoparticles [
13,
14,
15,
16,
17]. Even though there are reports on increased myricetin bioavailability that is encapsulated in microemulsions, prepared from oil, water, surfactant and co-surfactant [
18], and solid lipid nanoparticles (SLNs) [
19], there exist many drawbacks, which warrants the development of new approaches for myricetin encapsulation. Microemulsions are responsive to temperature and salinity changes and may undergo phase changes when exposed to higher- or lower-than-normal temperatures or salinity concentrations. This could lead to breaking of the microemulsion and, eventually, phase separation. Because of their crystalline structure and crystallization process, SLNs have drawbacks, such as low drug loading efficiency, possibilities of drug expulsion in storage conditions, and initial burst release.
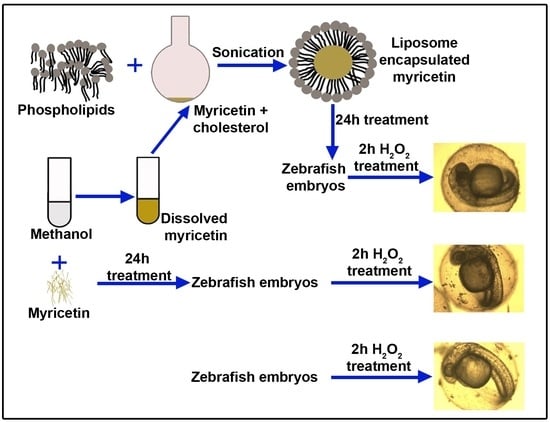
In this study, to offer improved nanoformulation, we encapsulated myricetin in liposomes and observed its antioxidant activity in zebrafish embryos after treatment with hydrogen peroxide (H2O2). H2O2 was used to induce oxidative stress in the embryos, which were pre-treated with only myricetin and nano-myricetin. Antioxidant enzyme activities, such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx), were assayed. To study the effect of myricetin on the developmental stages, we also observed the embryos microscopically and calculated the cumulative hatchability posttreatment with H2O2 and H2O2 pre-treated with only myricetin and nanoencapsulated myricetin. The work was executed to yield a superior product using liposomal nanoencapsulation that can be more biocompatible and bioavailable to exert its antioxidant effect.
2. Materials and Methods
2.1. Materials Used
Myricetin was purchased from TCI, Japan (Purity > 97.0%); ethanol, methanol, and chloroform were from E Merck; cholesterol, trypsin, collagenase, and DPPH were procured from HiMedia Laboratories Pvt Ltd.; and PBS tablets, sodium chloride (NaCl), ascorbic acid, dialysis membrane, and other chemicals were procured locally.
2.2. Isolation of Phospholipids from Egg Yolk
The yolk was collected from a hen egg, and phospholipids were isolated according to Ramesh et al. [
20] with slight modification. To 1 mL of egg yolk, 3 mL of 1 M NaCl was added and the mixture vortexed for proper mixing, followed by the addition of 12 mL of a methanol:chloroform mixture (2:1 ratio). The contents were mixed vigorously using a vortex. Further, 1 mL of 1 M NaCl and 3 mL of chloroform were added and the mixture vortexed. Later, this mixture was centrifuged at 2000 rpm for 5 min, which separated the mixture into three phases. The bottom organic phase was collected, which had pale-yellow-colored phospholipids, and was stored at −20 °C for thin-layer chromatography (TLC) confirmation.
2.3. Confirmation of Phospholipids by Thin-Layer Chromatography
TLC plates were activated after heating at 110 °C for 30 min. A line was drawn at the bottom, 0.5 cm above the edge, without scratching the gel and marked as the origin. With the help of a capillary tube, the extracted phospholipid was loaded at the center of the marked line and allowed to dry. Loading was repeated four times. The developing solvent was made by adding chloroform, methanol, and water in a ratio of 400:90:5 (i.e., chloroform:methanol:water = 8 mL:1.8 mL:0.1 mL) and placed in a beaker covered with a tight lid. After the loading of the sample was over, it was placed in the beaker and allowed to develop. When the solvent front reached near the top, the TLC plate was removed and the solvent front was marked. After drying in air, the TLC plates were placed in a large beaker containing iodine crystals and covered. After 15 min, the spots were stained in brown. The solvent front and sample spots were marked.
2.4. Nanoencapsulation of Myricetin
We used the procedure of Gaber et al. [
19] and Vimaladevi et al. [
21] for the nanoencapsulation of myricetin with slight modification. We added 5 µL of phospholipids, which were isolated from egg yolk, and 2 mg of cholesterol to an RB flask. To this, the methanol and chloroform mixture was added in a 1:3 ratio (2 mL of methanol:6 mL of chloroform). Then, 5 mg of myricetin, which was dissolved in 2 mL of methanol, was added to the RB flask. This complete mixture was evaporated by using a rotatory evaporator, and liposome-encapsulated myricetin was formed as a thin film inside the flask walls. Later, the RB flask with the thin film was kept under overnight incubation at room temperature in a desiccator for complete drying of the solvent. To prepare the liposome, the same process was executed without the addition of myricetin. The next day, we added 25 mL of PBS to the formed thin film in the RB flask and sonicated it for 20 min at room temperature. Then we filtered the sample with a 0.45 µm filter and stored it at 4 °C for further use. The characterization for size and stability was performed by dynamic light scattering using a Malvern zeta sizer. To explore the surface morphology of the liposome-encapsulated myricetin (n-Myr), we performed scanning electron microscopy (SEM). A thin layer of n-Myr was made above a solid support and dried in a dust-free atmosphere. Surface scanning was performed using JEOL JEM 2100 HRTEM with an accelerating voltage of 200 kV.
2.5. Drug Release Kinetics
The drug release kinetics of n-Myr was analyzed according to Deepika et al. [
22] using a dialysis membrane in a phosphate buffer saline (1X, pH 7.4) at room temperature. n-Myr (250 μM) was loaded in a dialysis membrane, and the membrane was placed in a beaker containing 50 mL of PBS solution (pH = 7.4). At specific time intervals, 2 mL of the released-n-Myr-containing buffer solution was withdrawn and the same volume of fresh PBS was added to maintain the total volume. The amount of released myricetin (Myr) was measured spectrophotometrically by measuring the absorbance at 324 nm. The encapsulation efficiency (EE) of n-Myr was evaluated spectrophotometrically [
22].
2.6. Free-Radical-Scavenging Activity
The free-radical-scavenging activity of Myr and n-Myr was determined by using DPPH assay with two different concentrations of Myr (50 µM and 250 µM) and n-Myr (50 µM and 250 µM). The free-radical-scavenging activity of the zebrafish embryo cell lysate was also assessed. Briefly, zebrafish embryos (10 embryos) were taken for each treatment group and treated with only liposomes without myricetin and individually with Myr-L, Myr-H, n-Myr-L, and n-Myr-H for 24 h. Untreated embryos were taken as controls. Treatment with H
2O
2 (3 mM) was performed after 24 h, and DPPH radical scavenging was carried out immediately after treatment and 2 h after treatment. Posttreatment, the embryos were converted into a single-cell suspension using a combination of trypsin and collagenase, as described by Bresciani et al. [
23]. Then the cells were lysed with lysis buffer (50 µM potassium phosphate buffer containing 0.5% Triton X-100) and the cell lysate (50 µL) was added to 2 mL of 100 µM DPPH. Absorbance was measured at 517 nm after 0 min and 30 min. Here, 50 mM ascorbic acid was taken as a positive control. The percentage of the free-radical-scavenging activity of cell lysate was measured according to the percentage of the inhibition formula [
24].
A0 = OD at 0th minute.
A30 = OD at 30th minute.
2.7. Culturing, Breeding of Zebrafish, and Collection of Embryos
Male and female zebrafish (
Danio rerio) were cultured in a rectangular fish tank according to Girigoswami et al. [
25] with slight modifications, by supplementing blood worms and flakes as food 2 times per day at room temperature (27 °C) in 12 h/12 h light/dark conditions. Care was taken, and the tank was cleaned once a week. To collect embryos, female and male zebrafish (3:4 ratio) were transferred into one fish tank in the evening and left for spawning. The next day, the embryos that were at the bottom of the tank were collected, washed with distilled water thrice, and used for the study.
2.8. Myricetin and Nanoformulated Myricetin Treatment
After collecting and washing the zebrafish embryos, we took 10–20 embryos in each well of the two 6-well plates. These wells we divided into 5 groups (control, myricetin low dose, myricetin high dose, nano-myricetin low dose, and nano-myricetin high dose) in duplicates (total 10 wells, 5 in one plate for treatment). The total volume of the solution in each well was 3 mL. We added 3 mL of distilled water (DIW) to the control group; then the final concentration of myricetin and nano-myricetin in low dose and high dose was 50 µM and 250 µM, adjusted with water and stock solutions of myricetin and nano-myricetin, respectively. The embryos were incubated for 20 h at room temperature (25 °C). Then the embryos were washed with distilled water twice, and all wells were filled with 3 mL of distilled water.
2.9. Hydrogen Peroxide Treatment
To observe the protective effect of myricetin and nano-myricetin against oxidative stress (OS), H2O2 was taken as a source of oxidant. After washing the embryos, we added 3 mM H2O2 to the myricetin- and nano-myricetin-treated embryos in each well to induce oxidative stress in the 6-well plate and incubated for 2 h at room temperature. After 2 h H2O2 treatment, the embryos were washed with distilled water twice and then filled with 3 mL of distilled water and incubated at room temperature. The embryos not treated with H2O2 were taken as a control.
2.10. Developmental Observations Using Microscopy
After H2O2 treatment, every day, embryos were analyzed under an inverted microscope. Due to the transparency of zebrafish embryos, we assessed the effect of myricetin and H2O2 on zebrafish embryos.
2.11. Cumulative Hatching
The cumulative hatchability was calculated, as performed previously [
25]. After the treatment, the embryos were incubated at room temperature and the number of hatched zebrafish embryos was noted in each well for 4 days. The embryos in each well at each time were calculated using the formula
A graph was created by taking time in hours postfertilization (hpf) on the abscissa and the percentage (%) of cumulative hatching on the ordinate according to Girigoswami et al. [
25].
2.12. Assay of Antioxidant Activities
Protein was estimated according to Lowry’s method. The antioxidant defense was assayed in H
2O
2-untreated and H
2O
2-treated zebrafish embryos that were pre-treated for 20 h with myricetin low dose (50 µm) and high dose (250 µm) and liposome-encapsulated myricetin low dose (50 µm) and high dose (250 µm). Antioxidant enzymes, such as CAT, GPx, and SOD, were assayed according to Girigoswami et al. [
2]. Both 2 h H
2O
2-treated and H
2O
2-untreated embryos were washed twice with distilled water and taken as respective controls. Calculations were carried out for antioxidant assays based on Girigoswami et al. [
2].
2.13. Catalase and Glutathione Peroxidase Assay
For catalase (CAT) and glutathione peroxidase assays from every group, we collected 10 embryos into the glass homogenizer and suspended them in lysis buffer (~300 µL) (50 µM potassium phosphate buffer containing 0.5% Triton X-100) and the homogenizer was kept on ice for 15 min for lysis of embryos. Then we homogenized the embryos with a piston by keeping the homogenizer on ice to maintain the temperature at 4 °C to hold enzyme activities until assay. We collected 50 µL of the lysate for protein estimation from each group of embryos. The protein estimation was performed with less volume of cell lysate (50 µL) by Lowry’s method (1951).
Then, the remaining extracted sample was centrifuged for 5 min at 800×
g at 4 °C, and the supernatant was taken for catalase assay by following the procedure of Claiborne [
26] and for glutathione peroxidase assay by following the method of Gunzler and Flohe [
27].
2.14. Superoxide Dismutase Assay
To assay the amount of SOD present in different cellular extracts, the SOD standard curve was drawn by plotting the amount of pure SOD (ng) vs. the percentage of NADH oxidation. The procedure followed was essentially the same as that by Paoletti et al. [
28]. A stock solution of standard SOD 100 µg/mL was prepared, and the amount was varied from 10 ng to 90 ng for the experiment. To find the SOD level, 10 zebrafish embryos were taken from each group and added to the homogenizer each time, and we added 600 µL of 100 mM TEA-DEA buffer (triethanolamine 665 µL from the stock of 1.12 kg/L, diethanolamine 480 µL from the stock of 1.091 kg/L, and HCl 690 µL (37%) were mixed and made up to 50 mL with d.H
2O; pH maintained at 7.4–7.5) and kept on ice for 15 min. Then the embryos were homogenized by keeping them on ice to avoid loss of enzyme activity. To remove low-molecular-weight substances, the supernatant was loaded into a Sephadex G-25 (coarse) column and sieved by centrifugation at 18,000 rpm for 5 min at 4 °C. The eluant contained the sample, which was immediately transferred to ice. From each sample, 3 µL of the supernatant was collected for protein estimation by Lowry’s method and the remaining supernatant was used for SOD assay by keeping it on ice until completion of SOD assay. Volumes from this supernatant were used for SOD measurement according to Paoletti et al. [
28].
2.15. Statistical Analysis
All the experiments were performed in triplicate. Student’s t-test was used to find the significance between the values.
4. Discussion
Myricetin is abundantly found in fruits and plants and in green tea, black tea, and wine. Myricetin plays a pivotal role in suppressing ROS and anticancer effects in vitro, but in vivo, due to its low solubility in water, it becomes difficult for it to reach the targeted cells [
5]. Nanotechnology has been used for nanoformulations to improve the bioavailability and drug efficacy, as well as being used in the detection of food toxins, biosensors, theranostics, imaging, etc. [
22,
29,
30,
31,
32,
33,
34,
35,
36]. Solid lipid nanoparticles have been used to encapsulate myricetin to improve its solubility, but they lack biocompatibility to some extent [
19]. Myricetin is found to be more potent in inhibiting glutathione reductase (GR) in the presence of O
2− than in the absence of it. Compared to other flavonols, a minimal concentration (110 µM) of myricetin inhibited 50% of GR [
37]. Myricetin increased GPx activity in Chinese hamster lung fibroblasts (V79-4) cells, as reported in an in vitro study [
38]. In DSS-induced colitis, murine showed increased GPx and SOD activities [
39]. The bioavailability of this polyphenol can be improved with the help of nanotechnology.
A nano-delivery system with a drug or nutraceuticals can better protect the drug from digestive enzymes as well as avoid the loss of the drug before it reaches the target and increase its bioavailability. Different types of nano-delivery systems are available based on the characteristics of a drug, such as liposome encapsulation, solid lipid particles (SLPs), nanostructured lipid carriers, self-dispersing lipid formulations, biopolymer-based delivery systems, and nano-laminated systems. Tang et al. reported myricetin and nano-myricetin encapsulated with Pluronic-based micelles and their effect on glioblastoma cells. It was found that nanoencapsulated myricetin induces higher cytotoxicity in glioblastoma cells than non-encapsulated myricetin [
40]. A myricetin nanoformulation with micelles improved ocular anti-inflammatory treatment and improved its stability and solubility [
41].
In this work, we used liposomes for the nanoencapsulation of myricetin for improved bioavailability, increased stability, and better solubility. Myricetin possesses needle-like structures when dissolved in PBS (
Figure S1), and it may cause damage to the cell wall when interacting with cells. To circumvent this needle-like morphology, it was necessary to encapsulate myricetin inside a nanostructure. The nanoencapsulation was carried out by synthesizing liposomes using the phospholipids isolated from egg yolk and cholesterol. The phospholipid isolation is shown in
Figure 1, where we can observe two bands, indicating phosphatidylcholine and phosphatidylethanolamine. Moreover, the absence of any other bands in the chromatogram, which can represent other impurities, such as free fatty acids, free cholesterol, cholesterol esters, or triglycerides, shows that the extraction was of pure phospholipids and there was no other component present in our extract. Previous reports indicate that the two bands in the chromatogram represent phosphatidylcholine and phosphatidylethanolamine and that other impurities (free fatty acids, free cholesterol, cholesterol esters, or triglycerides) are shown in other bands [
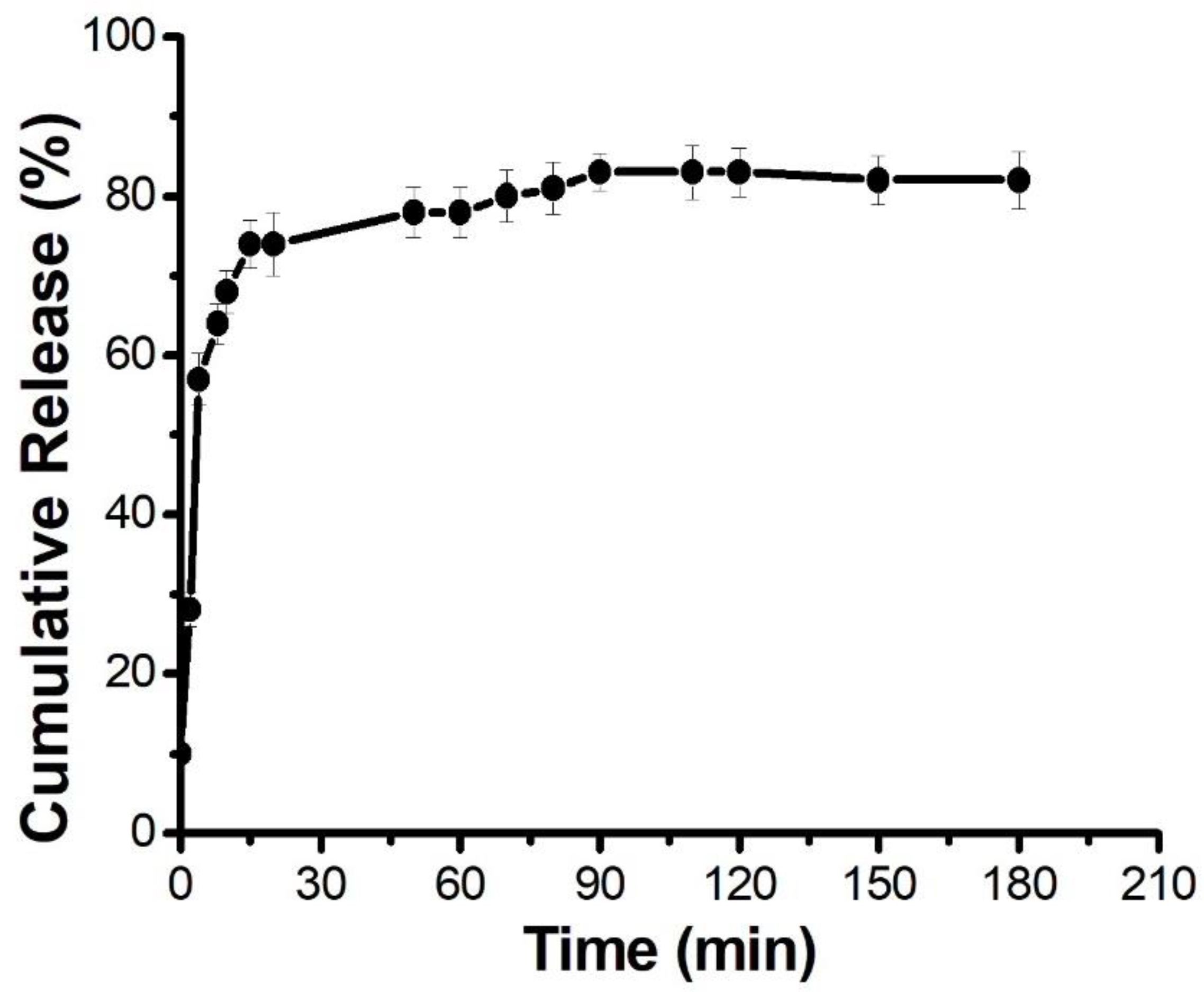
20]. The drug release kinetics of n-Myr was observed spectrophotometrically (
Figure 2). Initially, there was a burst release (up to 30 min), followed by slow release (up to 90 min). After 90 min, myricetin was released in a sustained manner. The encapsulation efficiency of n-Myr was found to be 72%. The synthesized liposomes were observed under a microscope (
Figure S2), which showed spherical liposomes. The liposomes were also stable, as visible from the zeta potential value of −30.6 mV (
Table 1). The liposome-encapsulated myricetin is shown in
Figure S3, where the absence of needle-like structures was observed. The characterization of nano-myricetin was performed using dynamic light scattering, and the results showed that the hydrodynamic diameter of the liposome-encapsulated myricetin was 262 nm, which is in the range defined for nanostructures. The particle was also highly stable, as indicated by the high-magnitude zeta potential of −20.2 mV (
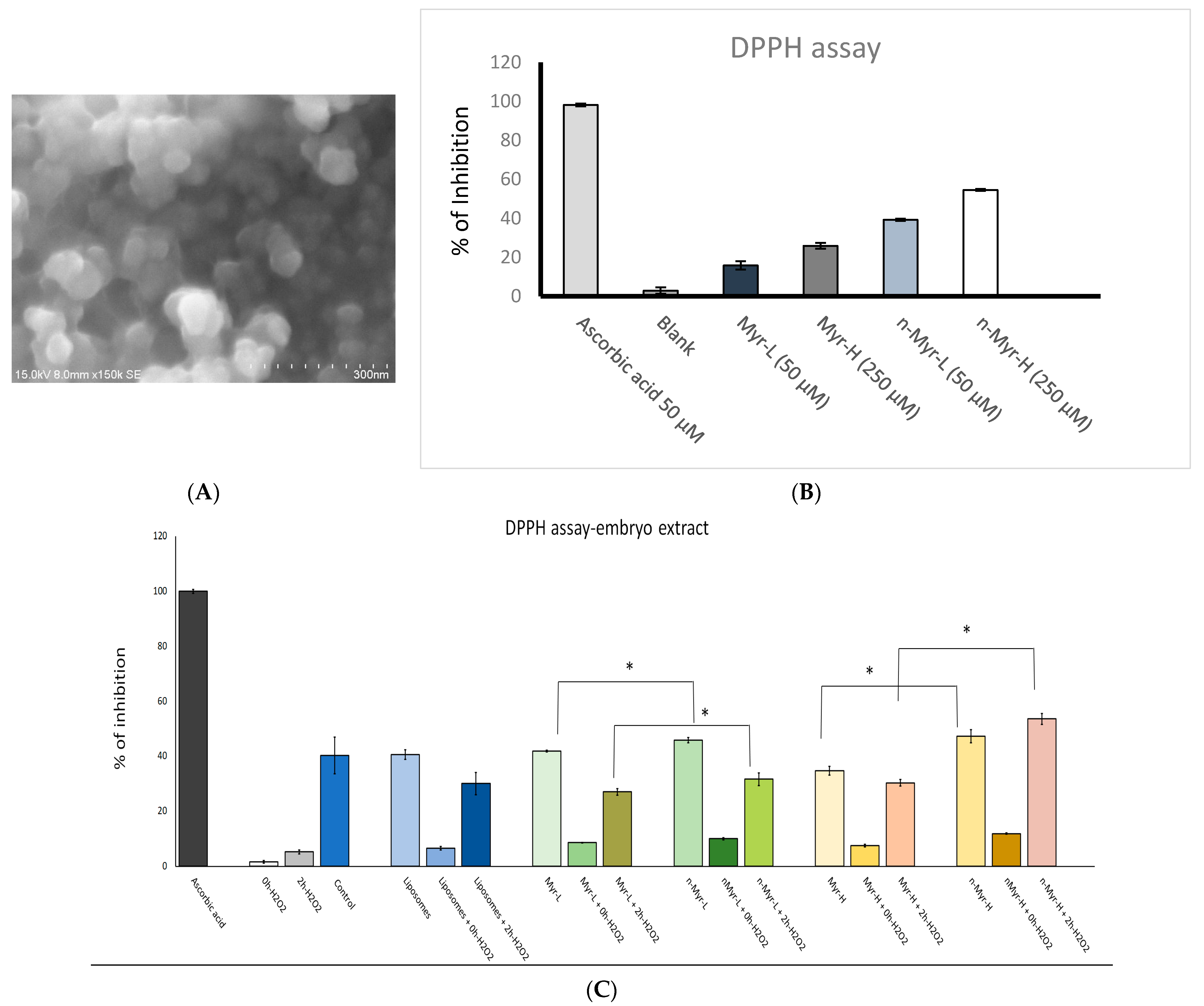
Table 1). The surface morphology was visualized by SEM imaging, which showed the spherical nature of the liposomal-encapsulated myricetin, with a diameter of 90 nm to 120 nm (
Figure 3A). Thus, the liposomal nanoformulation of myricetin was executed successfully. To establish the free-radical-scavenging activity, a DPPH assay was performed for Myr and for n-Myr. The results showed (
Figure 3B) that the free radical scavenging was 39.24% and 54.55% for a low dose and a high dose of n-Myr, respectively. The percentage inhibition was 15.81% and 25.84% for only Myr, respectively. Thus, for each dose, n-Myr exhibited higher free-radical-scavenging activity compared to only Myr. Further, we measured the total antioxidant capability of the embryos pre-treated with low and high doses of Myr and n-Myr (
Figure 3C) and further treatment with H
2O
2. Immediately after H
2O
2 treatment, there was a huge reduction (1.6%) in the radical-scavenging activity in cells that were not treated with Myr or n-Myr. However, 2 h after treatment, there was some improvement (5.2%) in radical-scavenging activity in cells that were not treated with either Myr or n-Myr. Cells untreated with H
2O
2 or Myr or n-Myr showed similar radical-scavenging activity as that of cells treated with only liposomes, as 40.27% and 40.58%, respectively. The immediate effect of H
2O
2 treatment and the effect 2 h posttreatment was observed for the Myr- and n-Myr-treated embryos, and the results demonstrate that immediate exposure to H
2O
2 caused a reduction in the percentage DPPH inhibition, which resumed after 2 h of treatment. The value returned to nearly that of control cells, but the radical-scavenging activity was significantly higher for all doses of n-Myr compared to Myr.
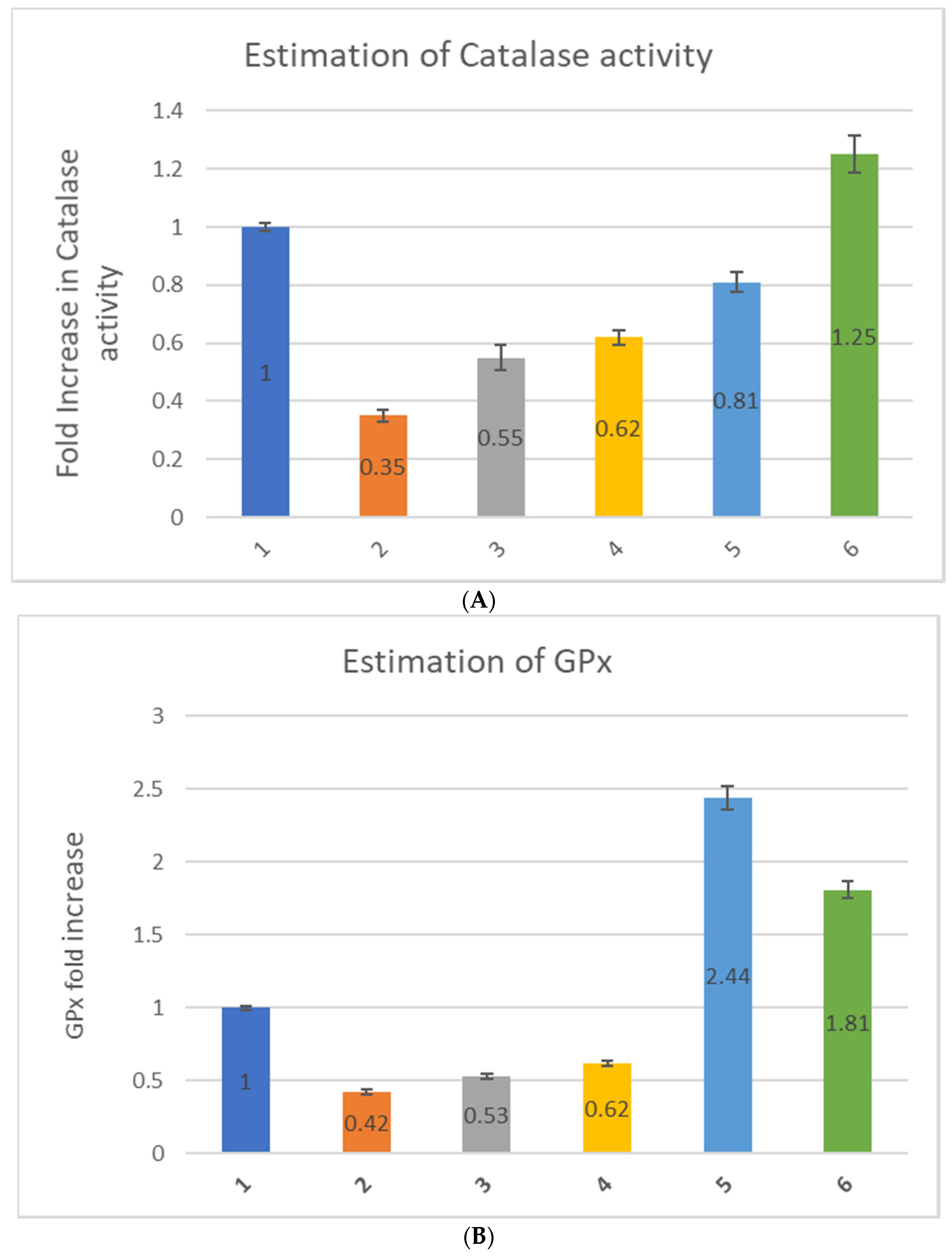
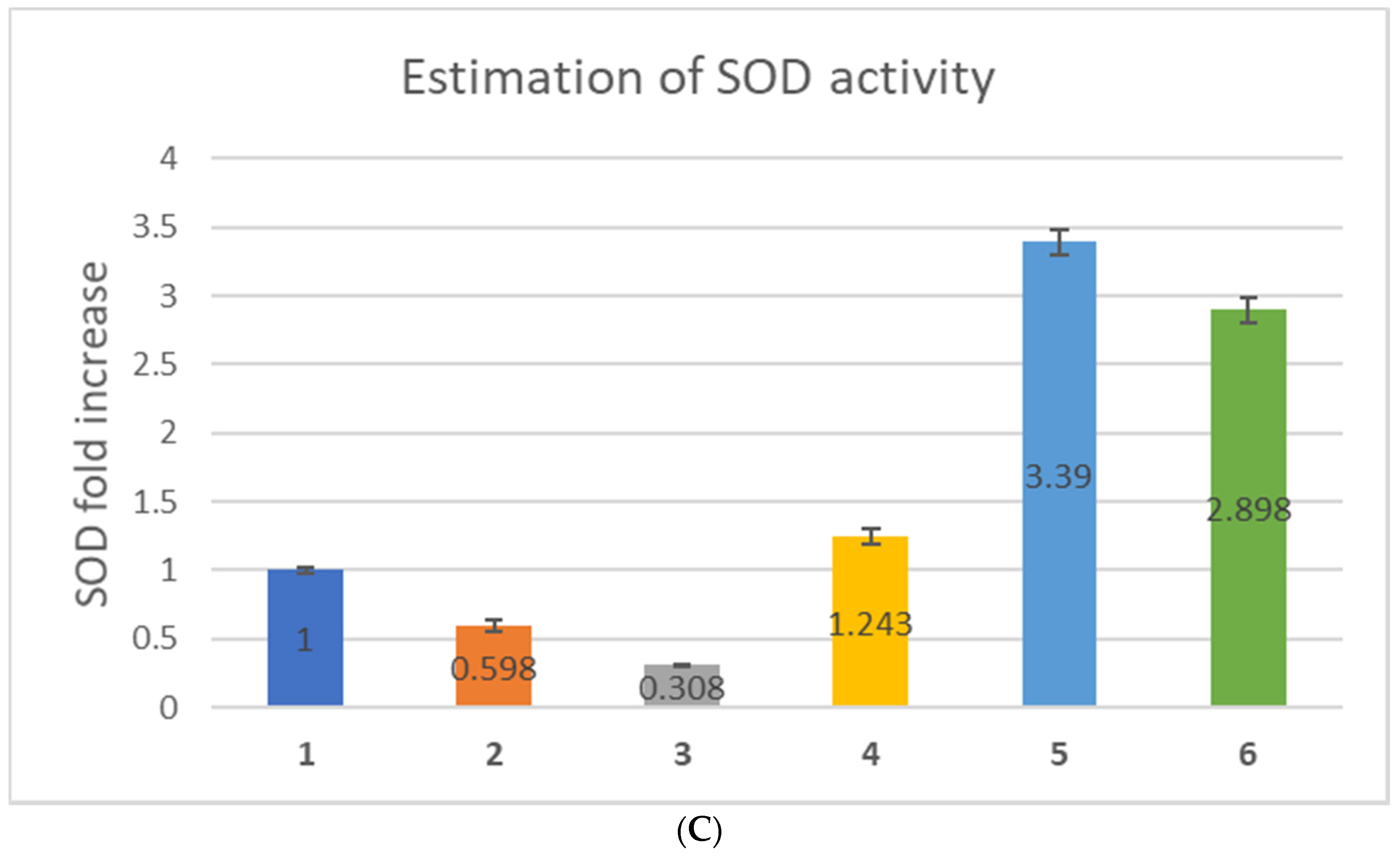
Further, to explore the effect of only myricetin and nano-myricetin on the antioxidant status of zebrafish embryos, we estimated the antioxidant enzyme activities, such as CAT, GPx, and SOD in zebrafish embryos. CAT activity decreased due to hydrogen peroxide treatment (0.35-fold), which increased to some extent after myricetin treatment. The increase was 0.55-fold after low-dose and 0.62-fold after high-dose treatment with myricetin (
Figure 4A). The enhancement was not that pronounced that it could bring back the catalase value similar to that of the control (1-fold). H
2O
2 treatment for n-Myr-L-pre-treated embryos showed improvement in CAT activity nearly similar to control embryos (0.81-fold), and interestingly there was a 25% increase in CAT activity found for n-Myr-H treatment. Thus, nanoencapsulated myricetin can offer protection against oxidative stress by elevating CAT activity. The results of GPx activity were also found to be fascinating. After treatment with hydrogen peroxide, there was a decrease in GPx activity (0.42-fold), which improved after treatment with Myr-L and Myr-H to 0.53- and 0.62-fold, respectively. However, after H
2O
2 exposure to liposome-encapsulated myricetin, there was an increase in GPx activity compared to both Myr-L- and Myr-H-treated zebrafish embryos (
Figure 4B). There was a 2.44-fold increase in the GPx level after n-Myr-L treatment and a 1.81-fold increase after treatment with n-Myr-H, showing a huge improvement in GPx activity induced by nanoencapsulated myricetin. The SOD value decreased 0.598-fold after treatment with H
2O
2, which decreased further to 0.308-fold after treatment with Myr-L (
Figure 4C). After high-dose treatment with myricetin, the SOD value reverted to that of the control (1.243-fold). In the case of nanoencapsulated myricetin pre-treatment, there was huge protection offered for H
2O
2-exposed zebrafish embryos and the increase in the SOD value was 3.39- and 2.898-fold for n-Myr-L and n-Myr-H, respectively. The results of antioxidant activity showed that the antioxidant enzyme levels decreased after treatment with only myricetin compared to treatment with nanoencapsulated myricetin. Myricetin was found to be 300 times stronger than other flavonoids in inhibiting catalase activity [
42]. We have resolved this limitation with liposome-encapsulated myricetin. Low- and high-dose-nano-myricetin-treated embryos expressed more catalase activity after H
2O
2 treatment compared to only low- and high-dose-myricetin-treated zebrafish embryos. From our results, we can speculate that the nanoencapsulation of myricetin could offer better protection against oxidative stress compared to only myricetin.
A step forward, we wanted to monitor the hatching ability of the zebrafish embryos after treatment with H
2O
2 and of myricetin- and nano-myricetin-pre-treated embryos further exposed to H
2O
2. The zebrafish embryos were pre-treated with myricetin as well as nano-myricetin at low and high doses and further exposed to H
2O
2. The images of the zebrafish embryos are shown in
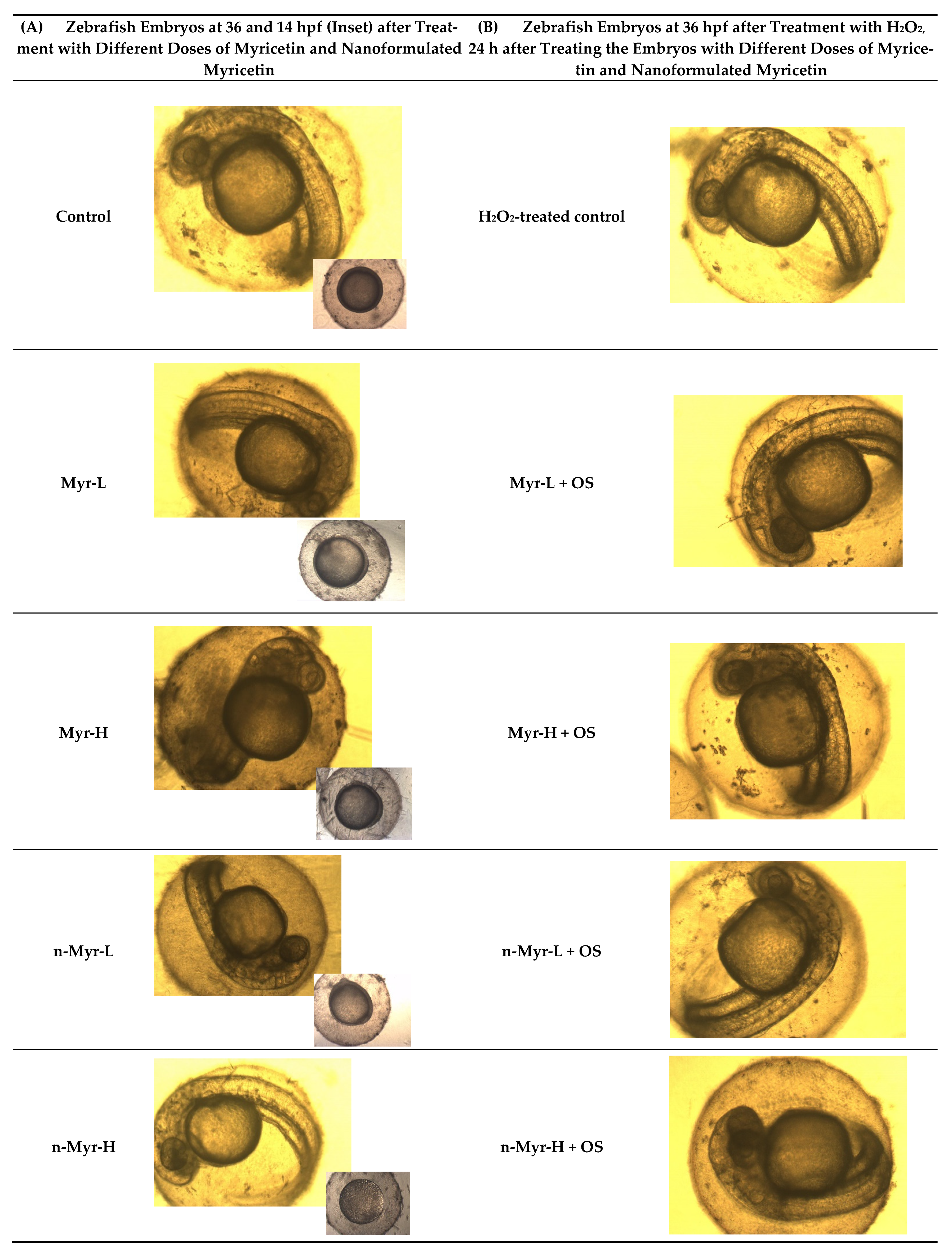
Figure 5, and we observed that at 14 hpf, there was an accumulation of needle-shaped myricetin particles on the embryo surface (
Figure 5A inset) and also that at 36 hpf, the medium for myricetin-treated samples was more turbid compared to that for samples treated with nanoformulated myricetin. Further, on treatment with H
2O
2, it was revealed that there was a deposition of some particles on the myricetin-treated embryos, which could block the supply of essential nutrients required for the survival of the embryos (
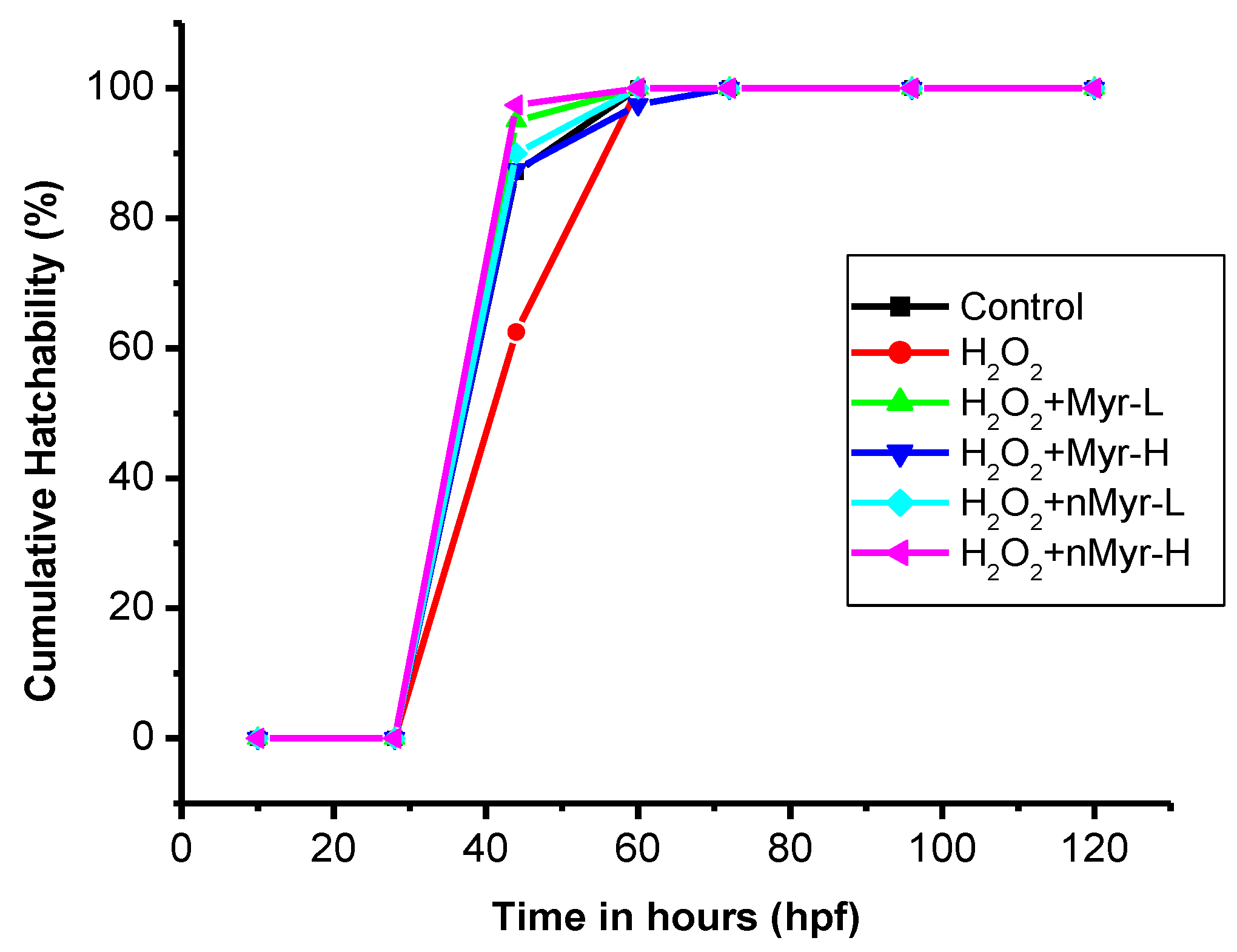
Figure 5B). There were no such deposits observed for nano-myricetin-treated embryos. The cumulative hatchability was also calculated for the hydrogen-peroxide-treated embryos (
Figure 6). The results showed that after 44 h, hatchability was 87.1% for untreated controls; 62.5% for H
2O
2-treated embryos; and 95% for Myr-L, 87.5% for Myr-H, 90% for n-Myr-L, and 97.4% for n-Myr-H doses. The results of cumulative hatchability showed that after H
2O
2 treatment, there was delayed hatching, whereas after treatment with myricetin as well as nano-myricetin, the hatching improved and the values were higher than those of the untreated control. When compared at a high dose, n-Myr showed 97.4% hatchability, whereas Myr showed 87.5%, which was significantly lower. After 60 h, there was 100% hatchability observed for all the groups except Myr-H, which showed 97.5% hatchability. Thus, it was observed that at a high dose, the hatchability was delayed for myricetin compared to nano-myricetin and the reason could be the aggregation of myricetin on the eggs, which hinders the exchange of essential nutrients and gases required for the growth and development of the embryos. However, the nanoformulation did not induce any aggregation and the embryos could hatch. A similar finding was observed for ZnO nanoparticles and chitosan-coated ZnO nanoparticles, which showed that ZnO nanoparticle treatment at high doses could induce aggregation on the top layer of the embryos and delay hatching, whereas chitosan coating does not cause any aggregation [
24]. Thus, the nanoformulation could protect the embryos from the needle-shaped myricetin particles (
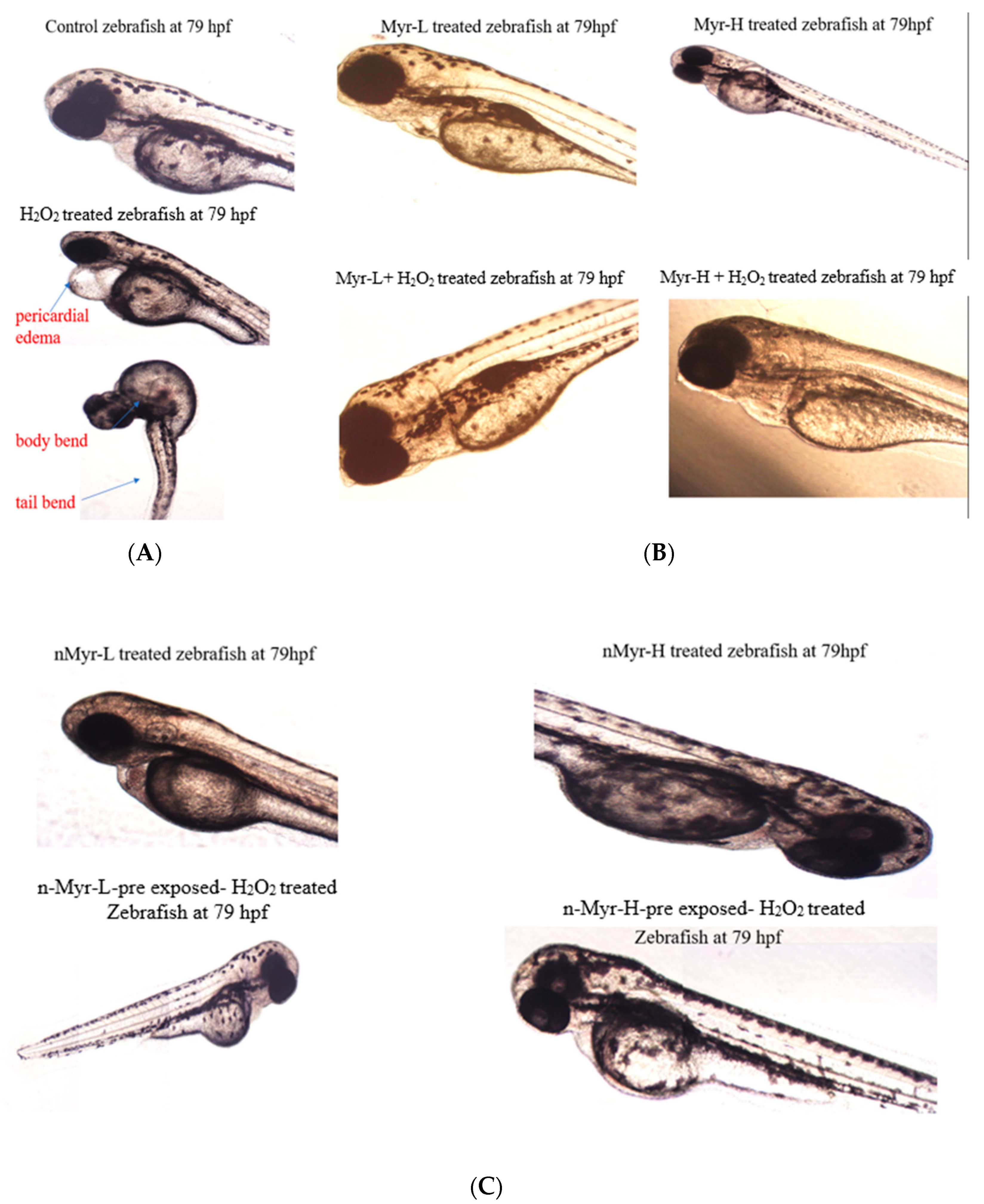
Figure S1). Posthatching, we could observe certain developmental abnormalities in the embryos, which are summarized in
Figure 7. H
2O
2-treated embryos developed abnormalities in their structures, such as pericardial edema, tail bend, and body bend. In other groups of embryos, such abnormalities were not observed post hatching. The findings from the above study show that liposomal nanoencapsulation of myricetin can improve the antioxidant status and is more biocompatible [
43]. Thus, nano-myricetin can impart a superior antioxidant effect as well as enable the healthy development of zebrafish embryos compared to myricetin.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}