Preparation and Chiral HPLC Separation of the Enantiomeric Forms of Natural Prostaglandins

Abstract
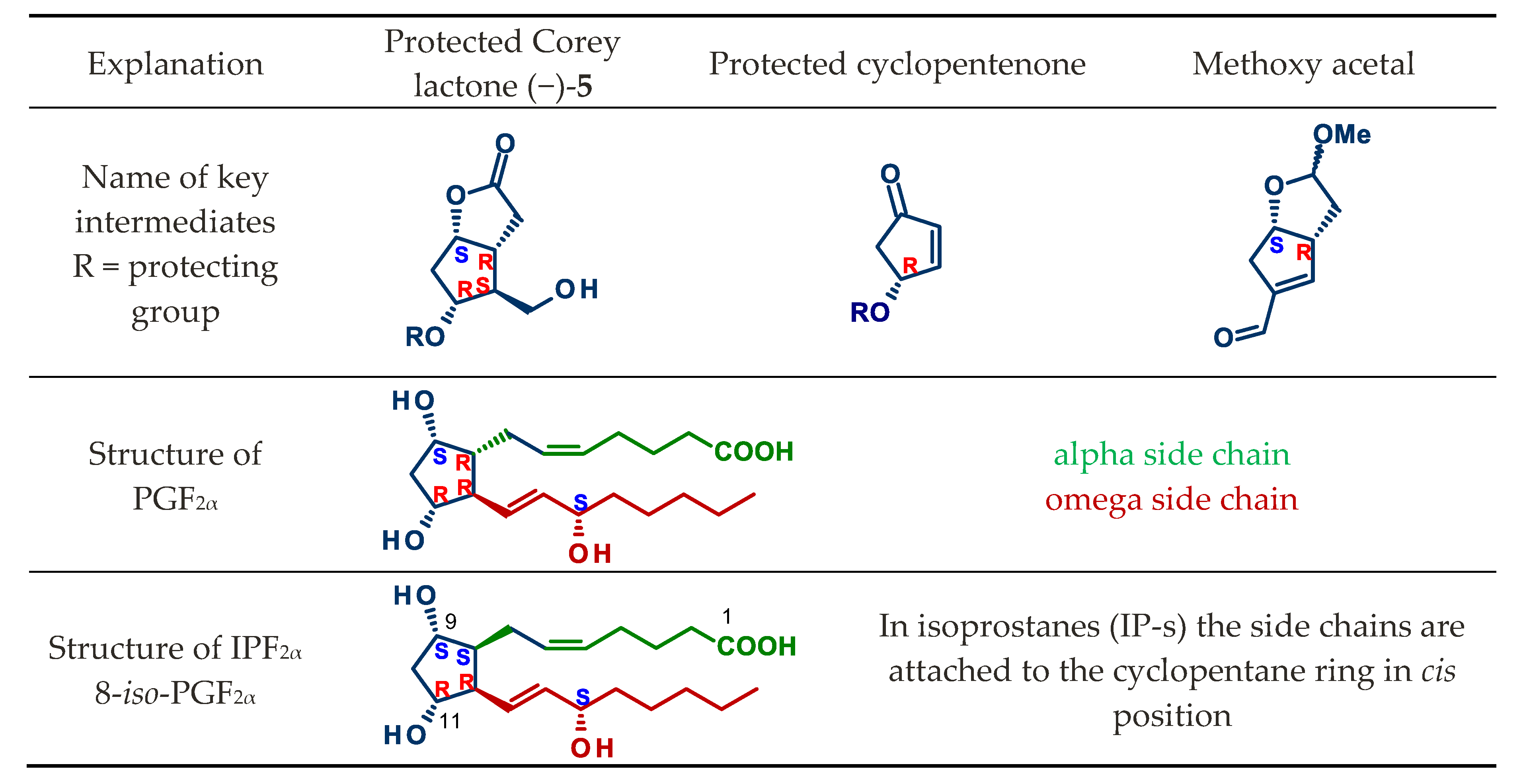
:1. Introduction
2. Aim of the Work
3. Material and Methods
4. Results and Discussions
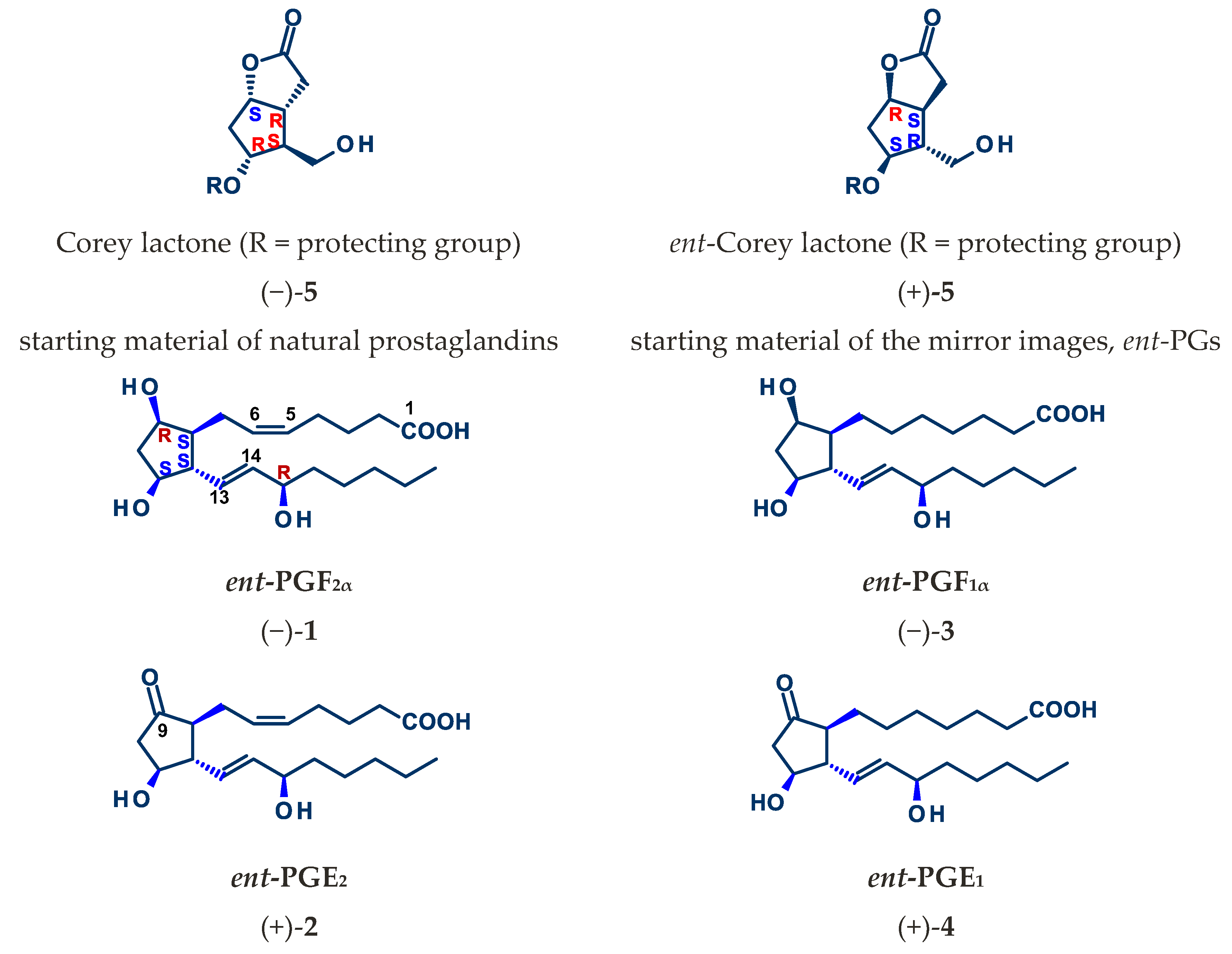
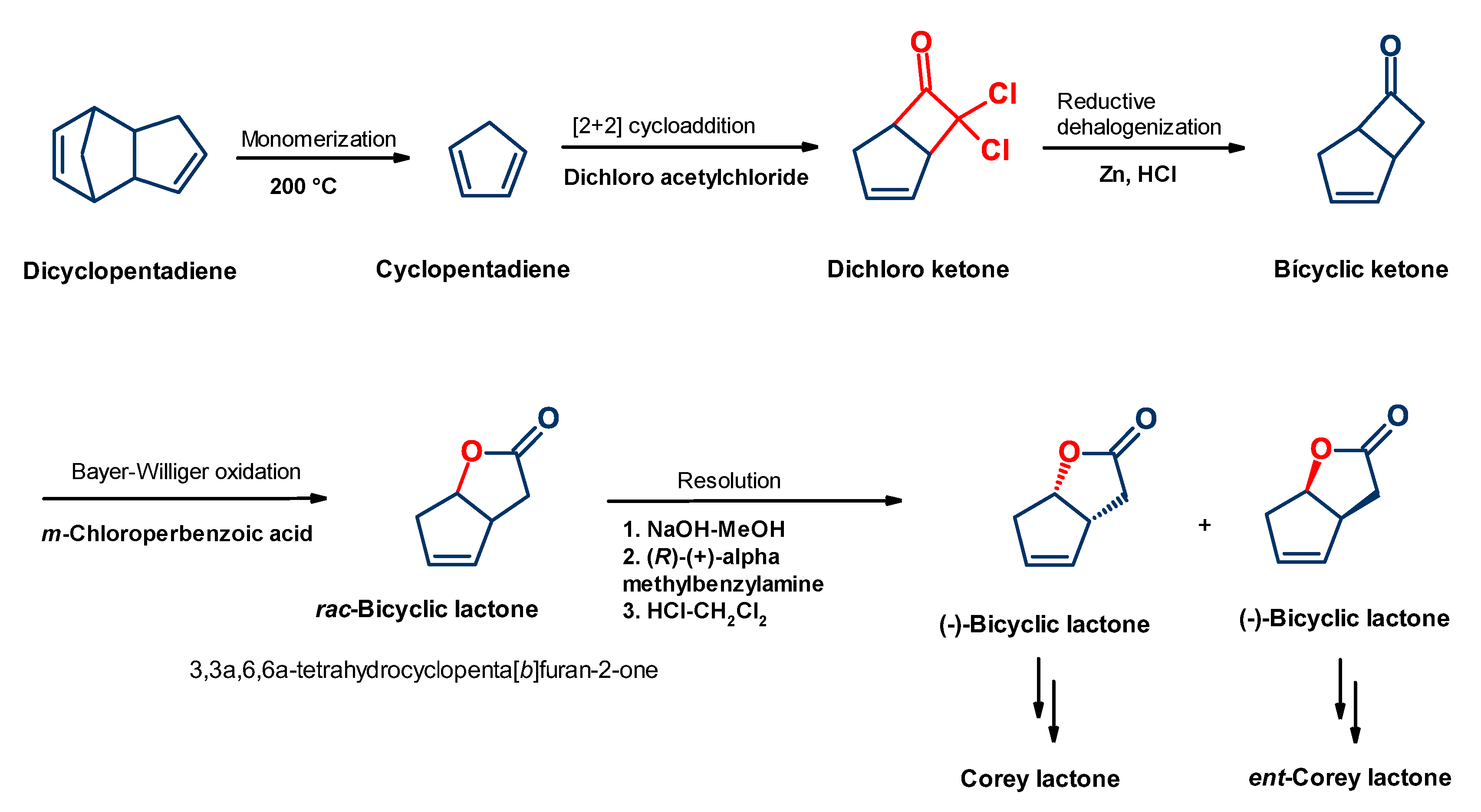
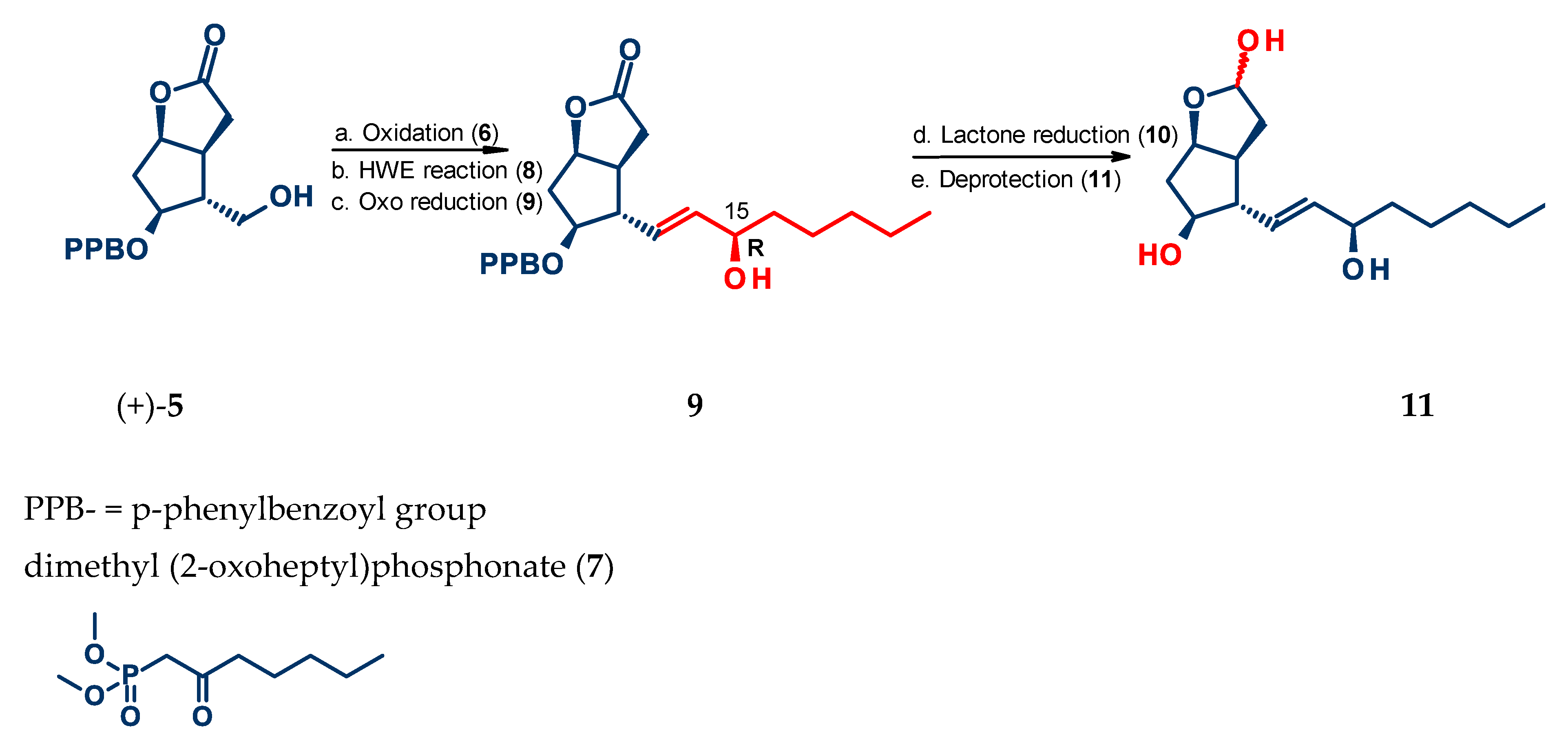
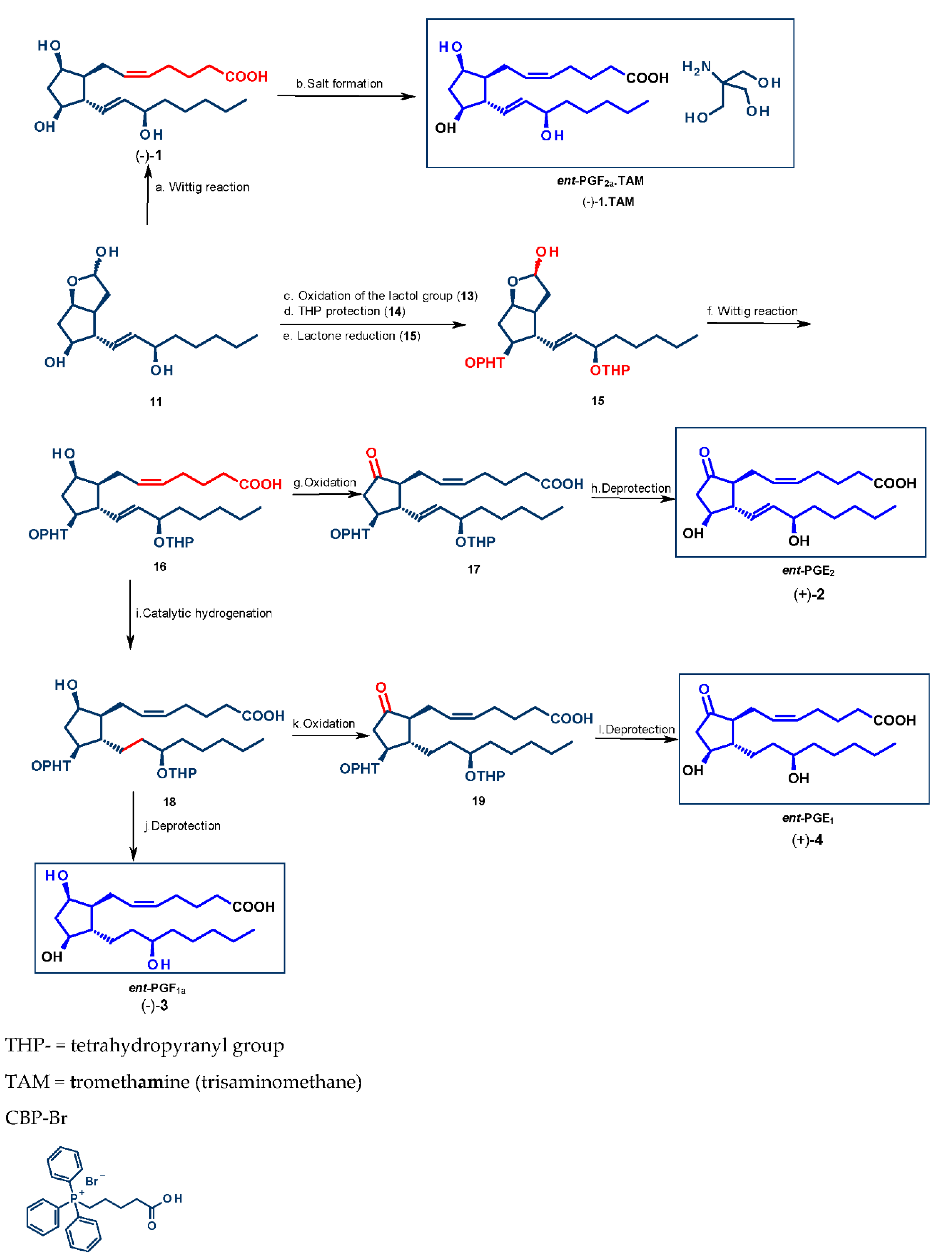
4.1. Synthesis of Mirror Images of Natural Prostaglandins
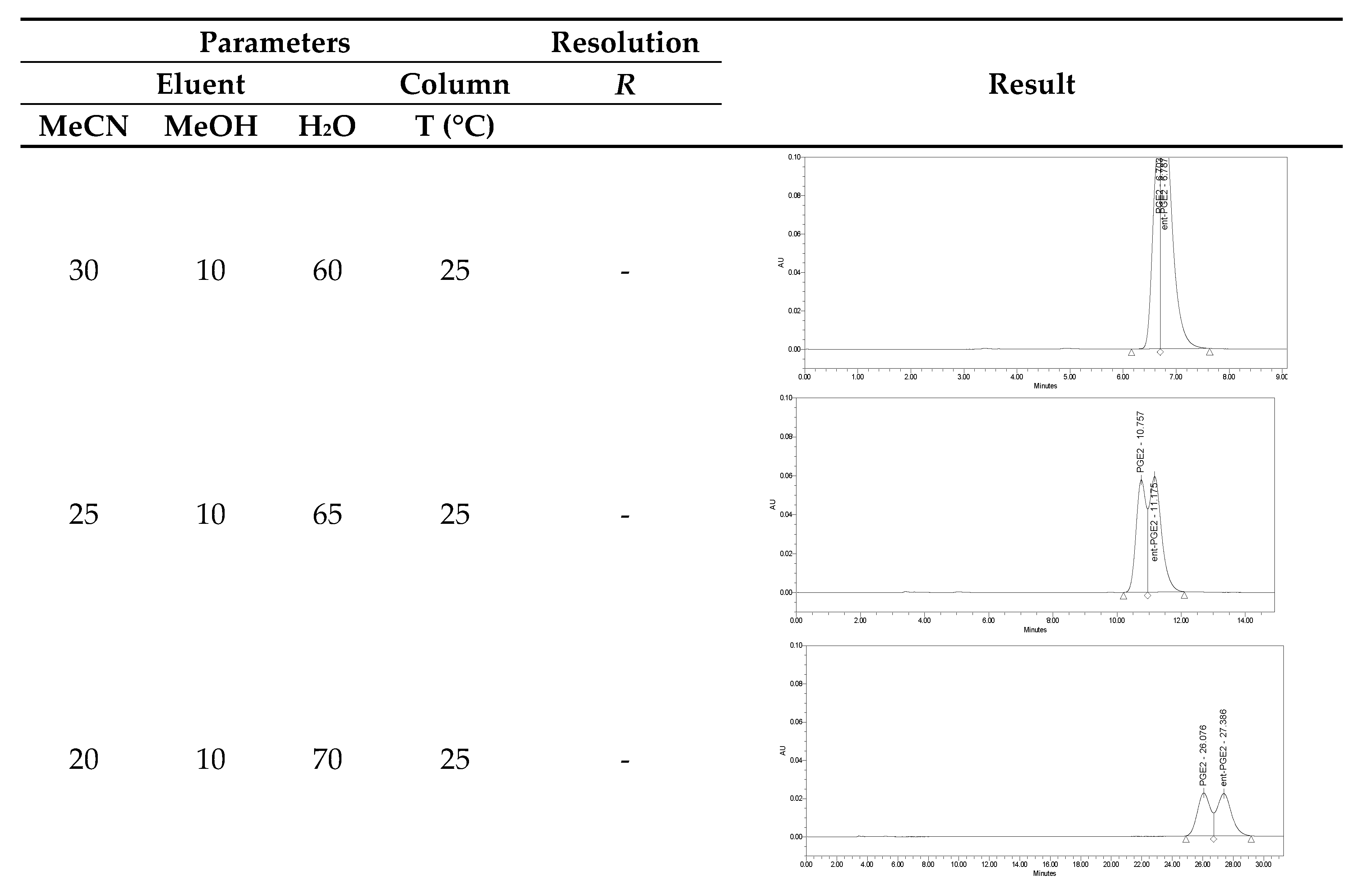
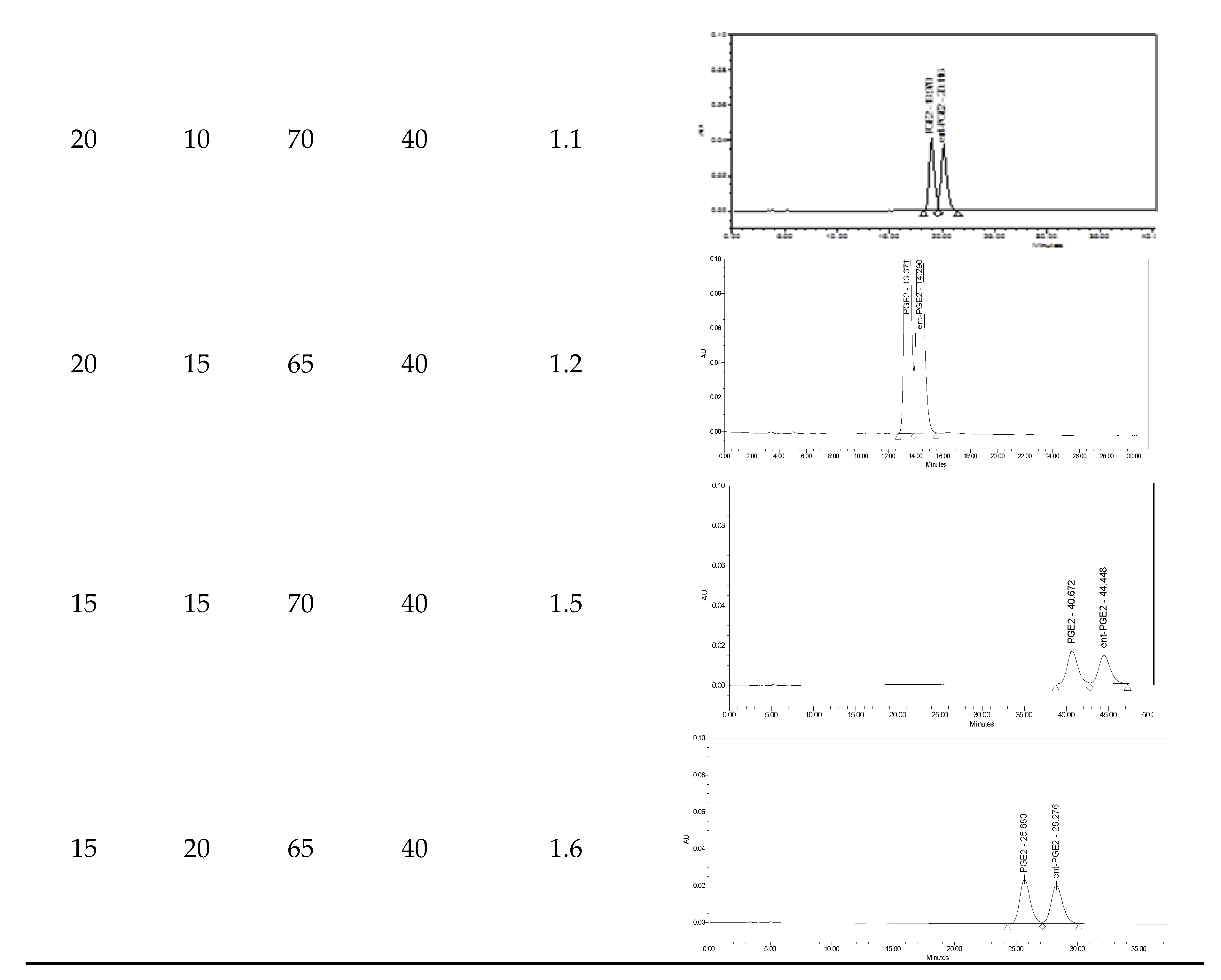
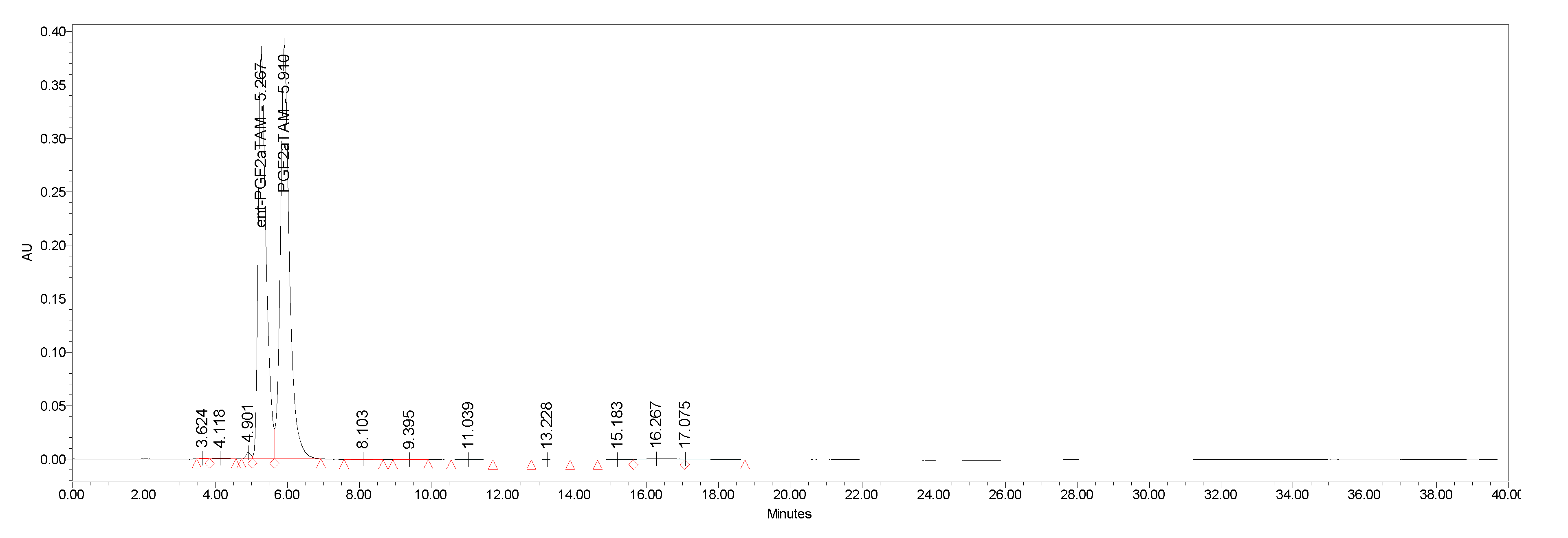
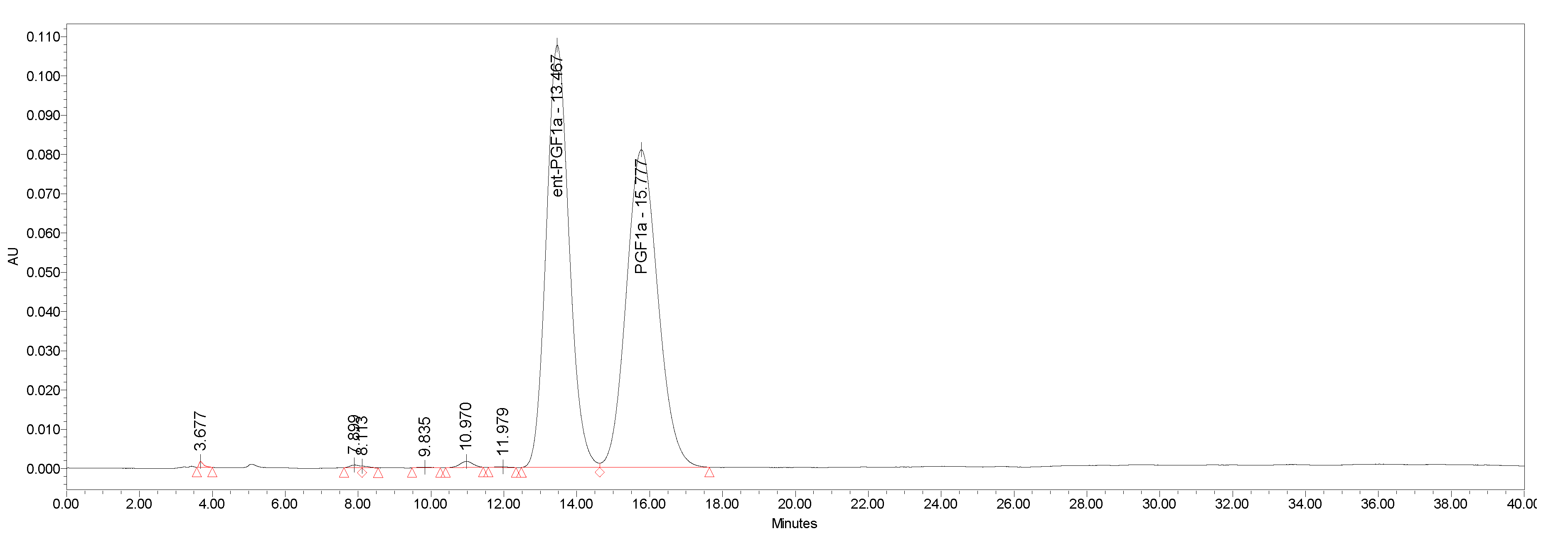
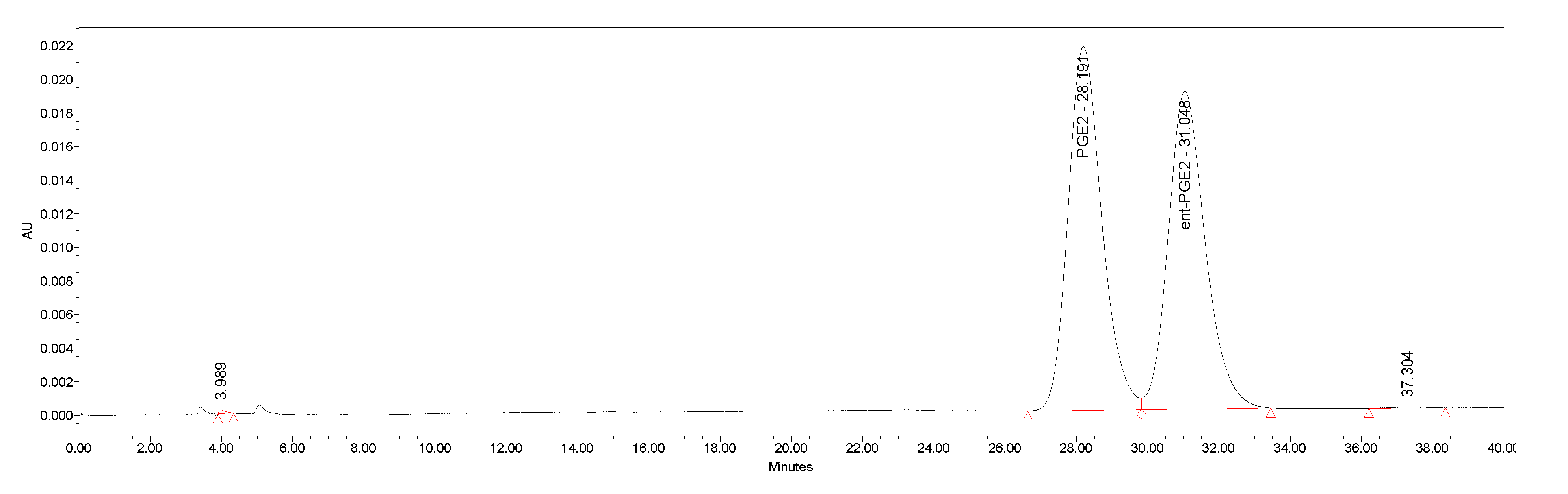
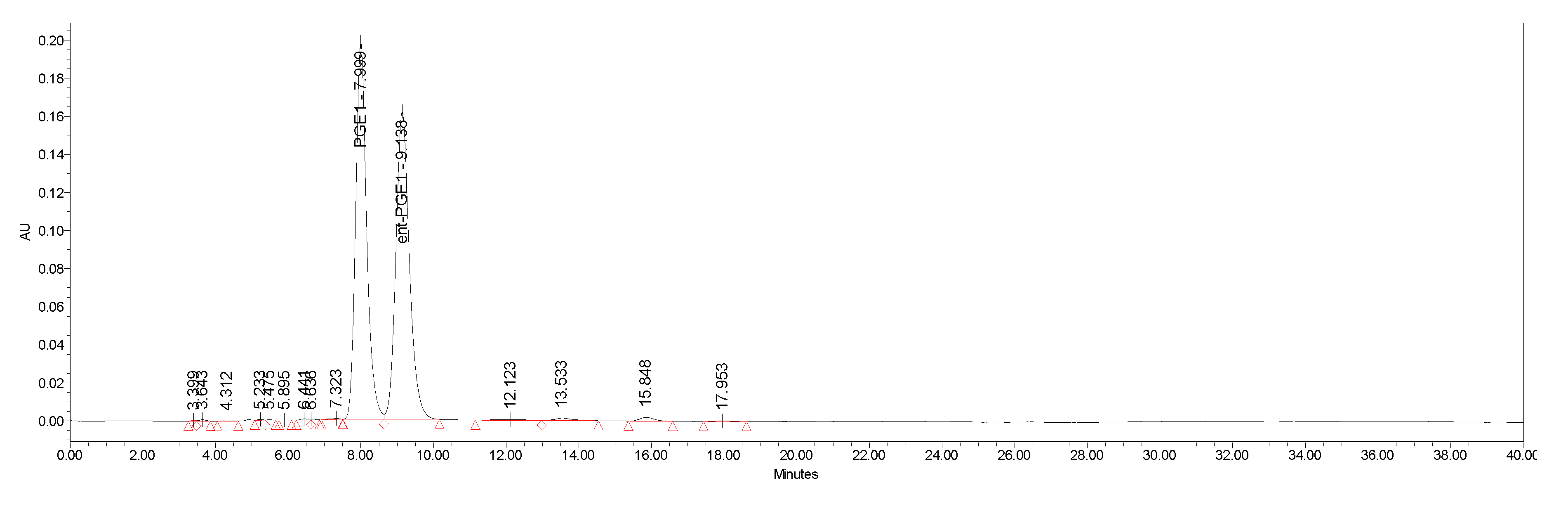
4.2. Separation of Natural Prostglandin Enantiomers by Chiral HPLC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bergström, S. The prostaglandins: From the Laboratory to the Clinic (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1983, 22, 858–866. [Google Scholar] [CrossRef]
- Samuelsson, B. From studies of biochemical mechanism to novel biological mediators: Prostaglandin endoperoxides, thromboxanes, and leukotrienes (Nobel lecture). Angew. Chem. Int. Ed. Engl. 1983, 22, 805–815. [Google Scholar] [CrossRef]
- Vane, J.R. Adventures and Excursions in bioassay: The steppingstones to prostacyclin (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1983, 22, 741–752. [Google Scholar] [CrossRef]
- Kurzok, R.C.; Lieb, C. Biochemical studies of human semen. II. The action of semen on the human uterus. Proc. Soc. Exp. Biol. Med. 1930, 28, 268–272. [Google Scholar] [CrossRef]
- Goldblatt, M.W. Properties of human seminal plasma. J. Phisyol. 1935, 84, 208–218. [Google Scholar] [CrossRef] [PubMed]
- von Euler, U.S. On the specific vasodilating and plain muscle stimulating substances from accessory genital glands in man and certain animals (prostaglandin and vesiglandin). J. Phisyol. 1937, 88, 213–234. [Google Scholar]
- Corey, E.J. The logic of chemical synthesis: Multistep synthesis of complex carbogenic molecules (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1991, 30, 455–465. [Google Scholar] [CrossRef]
- Corey, E.J.; Weinshenker, N.M.; Schaaf, T.K.; Huber, W. Stereo-controlled synthesis of prostaglandins F2a and E2 (dl). J. Am. Chem. Soc. 1969, 91, 5675–5677. [Google Scholar] [CrossRef]
- Corey, E.J.; Schaaf, T.K.; Huber, W.; Koelliker, U.; Weinshenker, N.M. Total synthesis of prostaglandins F2a and E2 as the naturally occurring forms. J. Am. Chem. Soc. 1970, 92, 397–398. [Google Scholar] [CrossRef]
- Corey, E.J.; Noyori, R.; Schaaf, T.K. Total synthesis of prostaglandins F1a, E1, F2a, and E2 (Natural forms) from a common synthetic intermediate. J. Am. Chem. Soc. 1970, 92, 2586–2587. [Google Scholar] [CrossRef]
- Sih, C.J.; Salomon, R.G.; Price, P.; Peruzzotti, G.; Sood, R.G. Total synthesis of (±)-15-deoxyprostaglandin E1. J. Chem. Soc. Chem. Commun. 1972, 4, 240–241. [Google Scholar] [CrossRef]
- Sih, C.J.; Price, P.; Sood, R.; Salomon, R.G.; Peruzzotti, G.; Casey, M. Total synthesis of prostaglandins II. Prostaglandin E1. J. Am. Chem. Soc. 1972, 94, 3643–3644. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Suzuki, M. Prostaglandin synthesis by tree-component coupling. Angew. Chem. Int. Ed. Engl. 1984, 23, 847–876. [Google Scholar] [CrossRef]
- Suzuki, M.; Morita, Y.; Koyano, H.; Koga, M.; Noyori, R. Three-component coupling synthesis of prostaglandins. A simplified, general procedure. Tetrahedron 1990, 46, 4809–4822. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Coulthard, G.; Erb, W.; Aggarwal, V.K. Stereocontrolled organocatalytic synthesis of prostaglandin PGF2a in seven steps. Nature 2012, 489, 278–281. [Google Scholar] [CrossRef]
- Baars, H.; Classen, M.J.; Aggarwal, V.K. Synthesis of Alfaprostol and PGF2a through 1,4-addition of an alkyne to an enal intermediate as the key step. Org. Lett. 2017, 19, 6008–6011. [Google Scholar] [CrossRef] [Green Version]
- Pelss, A.; Gandhamsetty, N.; Smith, J.R.; Mailhol, D.; Silvi, M.; Watson, A.J.A.; Perez-Powell, I.; Prévost, S.; Schützenmeister, N.; Moore, P.R.; et al. Reoptimization of the organocatalyzed double aldol domino process to a key enal intermediate and its application to the total synthesis of delta12 prostaglandin J3. Chem. Eur. J. 2018, 24, 9542–9545. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.W.; Djuric, S.W. Synthesis of therapeutically useful prostaglandin analogs. Chem. Rev. 1993, 93, 1533–1564. [Google Scholar] [CrossRef]
- Das, S.; Chandrasekhar, S.; Yadav, J.S.; Grée, R. Recent developments in the synthesis of prostaglandin analogues. Chem. Rev. 2007, 107, 3286–3337. [Google Scholar] [CrossRef]
- Peng, H.; Chen, F.-E. Recent advances in asymmetric total synthesis of prostaglandins. Org. Biomol. Chem. 2017, 15, 6281–6301. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.J. Prostaglandins in veterinary practice. In Pract. 1981, 3, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.J.; Hammond, D.; Schultz, R.H. Veterinary uses of prostaglandins. In Practical Applications of Prostaglandins and Their Synthesis Inhibitors, 1st ed.; Kerim, S.M.M., Ed.; Springer: Berlin/Heidelberg, Germany; MPP Press Ltd.: Dordrecht, The Netherlands, 1979; pp. 190–209. [Google Scholar]
- Lee, C.W.; Buckley, F.; Costello, S.; Kelly, S. A systematic review of the characteristics of randomised control trials featuring prostaglandins for the treatment of glaucoma. Curr. Med. Res. Opin. 2008, 24, 2265–2270. [Google Scholar] [CrossRef] [PubMed]
- Toris, C.; Gulati, V. The biology, pathology and therapeutic use of prostaglandins in the eye. Clin. Lipidol. 2011, 6, 577–591. [Google Scholar] [CrossRef]
- Lee, S.H.; Rubin, L.J. Current treatment strategies for pulmonary arterial hypertension. J. Internal Med. 2005, 258, 199–215. [Google Scholar] [CrossRef]
- Buckley, M.S.; Berry, A.J.; Kazem, N.H.; Patel, S.A.; Librodo, P.A. Clinical utility of treprostinil in the treatment of pulmonary hypertension: An evidence based review. Core Evid. 2014, 9, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, J.H.; Park, Y.S.; Suk, K.S.; Lee, J.H.; Park, M.S.; Moon, S.H. Comparative study of the efficacy of limaprost and pregabalin as single agents and in combination for the treatment of lumbar spinal stenosis: A prospective, double-blind, randomized controlled non-inferiority trial. Spine J. 2016, 16, 756–763. [Google Scholar] [CrossRef]
- Satoh, H.; Takeuchi, K. Management of NSAID/Aspirin-Induced Small Intestinal Damage by GI-Sparing NSAIDs, Anti-Ulcer Drugs and Food Constituents. Curr. Med. Chem. 2012, 19, 82–89. [Google Scholar] [CrossRef]
- Jozwiak, M.; Oude Rengerink, K.; Ten Eikelder, M.L.G.; van Pampus, M.G.; Dijksterhuis, M.G.K.; de Graaf, I.M.; van der Post, J.A.M.; van der Salm, P.; Scheepers, H.C.J.; Schuitemaker, N.; et al. Foley catheter or prostaglandin E2 inserts for induction of labour at term: An open-label randomized controlled trial (PROBAAT-P trial) and systematic review of literature. Eur. J. Obst. Gynecol. Reprod. Biol. 2013, 170, 137–145. [Google Scholar] [CrossRef]
- Distefano, G. The pharmacological manipulation of Botallo’s duct in the duct-dependent congenital cardiopathies and in the preterm infants with respiratory distress. A review and personal findings. Minerva Pediatrica 2005, 57, 21–34. [Google Scholar]
- Huang, F.K.; Lin, C.C.; Huang, T.C.; Weng, K.P.; Liu, P.Y.; Chen, Y.Y.; Wang, H.P.; Ger, L.P.; Hsieh, K.S. Reappraisal of the prostaglandin E1 dose for early newborns with patent ductus arteriosus-dependent pulmonary circulation. Pediatr. Neonat. 2013, 54, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.E. Lubiprostone: A new drug for the treatment of chronic idiopathic constipation. Rev. Gastroent. Disord. 2007, 7, 214–222. [Google Scholar]
- Corey, E.J.; Andersen, N.H.; Carlson, R.M.; Paust, J.; Vedejs, E.; Vlattas, I.; Winter, R.E.K. Total synthesis of prostaglandins. Synthesis of the pure dl-E1, -F1a, F1b, -A1 and -B1. J. Am. Chem. Soc. 1968, 90, 3247. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Vlattas, I.; Harding, K. Total synthesis of natural (levo) and enantiomeric (dextro) forms of prostaglandin E1. J. Am. Chem. Soc. 1969, 91, 535–536. [Google Scholar] [CrossRef]
- Slaters, H.L.; Zelawski, Z.S.; Taub, D.; Wendler, N.L. A new stereoselective total synthesis of prostaglandin E1 and its optical antipodes. J. Chem. Soc. Chem. Commun. 1972, 6, 304–305. [Google Scholar] [CrossRef]
- Slaters, H.L.; Zelawski, Z.S.; Taub, D.; Wendler, N.F. A stereoselective total synthesis of nat(−)-prostaglandin E1 and its optical antipode. Tetrahedron 1974, 30, 819–830. [Google Scholar] [CrossRef]
- Taub, D.; Hoffsommer, R.D.; Kuo, C.H.; Zelaeski, Z.S.; Wendler, N.L. A stereoselective total synthesis of PGE1. Chem. Comm. 1970, 1258–1259. [Google Scholar]
- Cooper, E.L.; Yankee, E.W. Enantiomeric prostaglandins. J. Am. Chem. Soc. 1974, 96, 5876–5894. [Google Scholar] [CrossRef]
- Andersen, N.H.; Imamoto, S.; Picker, D.H. Synthesis of bis-unsaturated prostaglandins. Prostaglandins 1977, 14, 61–101. [Google Scholar] [CrossRef]
- Ramwell, P.W.; Shaw, J.E.; Corey, E.J.; Andersen, N. Biological activity of synthetic prostaglandins. Nature 1969, 221, 1251–1253. [Google Scholar] [CrossRef]
- Miller, W.L.; Sutton, M.J. Relative biological activity of certain prostaglandins and their enantiomers. Prostaglandins 1976, 11, 77–84. [Google Scholar] [CrossRef]
- Morrow, J.D.; Harris, T.M.; Roberts, L.J., II. Non-cyclooxygenase oxidative formation of a series of novel prostaglandins: Analytical ramifications for measurement of eicosanoids. Anal. Biochem. 1990, 184, 1–10. [Google Scholar] [CrossRef]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., II. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, G.L.; Dai, Q.; Roberts, L.J., II. The isoprostanes 25 years later. Biochim. Biophis. Acta 2015, 1851, 433–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oge, K.; Cuyamendous, C.; de la Torre, A.; Candy, M.; Guy, A.; Bultel-Poncé, V.; Durand, T.; Galano, J.M. History of chemical routes towards cyclic non-enzymatic oxygenated metabolites of polyunsaturated fatty acids. Synthesis 2018, 50, 3257–3280. [Google Scholar]
- Antus, B.; Kardos, Z. Oxidative stress in COPD: Molecular background and clinical monitoring. Curr. Med. Chem. 2015, 22, 627–650. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Tömösközi, I.; Gruber, L.; Kovács, G.; Székely, I.; Simonidesz, V. Regiospecific Prins reaction, a new way to prostanoids. Tetrahedron Lett. 1976, 50, 4639–4642. [Google Scholar] [CrossRef]
- Szabó, T.; Dalmadi, G.; Remport, R.I.; Forró, L.; Hermecz, I.; Sárközi, P.; Nemes, J. Process for the Preparation of PGF2a Tromethamine Salt. Patent HU 56060 A2, 29 July 1991. [Google Scholar]
- Ivanics, J.; Szabó, T.; Hermecz, I.; Dalmadi, G.; Kovacs, G.; Resul, B. Chemical Process: Preparation of 13,14-dihydro-15(R)-17-phenyl-18,19,20-trinor-PGF2a Esters. Patent WO 1993000329 A1, 7 January 1993. [Google Scholar]
- Dalmadi, G.; Hajdu, F.; Hermecz, I.; Mozsolits, K.; Szabo, T.; Szeverényi, Z.; Vajda, E. Process for the Preparation of Prostaglandin E1. Patent WO 1997022881 A1, 26 June 1997. [Google Scholar]
- Galambos, G.; Benno, N.; Povarny, B.M.; Dalmadi, G.; Ebinger, K.; Forró, L.; Hajdu, F.; Hermecz, I.; Kardos, Z.; Lukacs, G.; et al. Process for the Selective Reduction of α,β-Unsaturated Ketones as Prostaglandin Intermediates. Patent WO 1997022602 A2, 26 June 1997. [Google Scholar]
- Szabó, T.; Bódi, J.; Dalmadi, G.; Kardos, Z.; Szeverényi, Z. Process for the Preparation of Beraprost and Its Salts. Patent WO 2003011849 A1, 13 February 2003. [Google Scholar]
- Kardos, Z.; Szeverényi, Z.; Dalmadi, G.; Szabó, T.; Bódi, J. Production of Beraprost Ester by Selective reduction. Patent HU 0103088 A2, 28 July 2001. [Google Scholar]
- Havasi, G.; Kiss, T.; Hortobágyi, I.; Kardos, Z.; Lászlófi, I.; Bischof, Z.; Bódis, Á. Novel Processes for the Preparation of Prostaglandin Amides. Patent WO 2012164324 A1, 6 December 2012. [Google Scholar]
- Kardos, Z.; Kiss, T.; Lászlófi, I.; Hortobágyi, I.; Bischof, Z.; Bódis, Á.; Havasi, G. Process for the Preparation of Travoprost. Patent WO 2013093528 A1, 27 June 2013. [Google Scholar]
- Bischof, Z.; Bódis, Á.; Kőműves, M.M. Alternative Process for the Preparation of Travoprost. Patent WO 2014083367 A1, 5 June 2014. [Google Scholar]
- Vajda, E.; Hortobágyi, I.; Lászlófi, I.; Buzder-Lantos, P.; Havasi, G.; Takács, L.; Kardos, Z. New Process for the Preparation of High Purity Prostaglandins. Patent WO 2015136317 A1, 17 September 2015. [Google Scholar]
- Juhász, I.; Hortobágyi, I.; Altsach, T.; Lászlófi, I.; Borkó, N.Á.; Rozsumberszki, I.; Havasi, G.; Kardos, Z.; Buzder-Lantos, P. Process for the Preparation of Treprostinil and Its Salt Forms. Patent WO 2016055819 A1, 14 April 2016. [Google Scholar]
- Buzder-Lantos, P.; Kardos, Z.; Hortobágyi, I.; Lászlófi, I.; Juhász, I.; Fónagy, L.; Váradi, C.; Borkó, N.Á. Process for the Preparation of Carboprost and Its Tromethamine Salt. Patent WO 2017093770 A1, 8 June 2017. [Google Scholar]
- Takács, L.; Fekete, I.; Buzder-Lantos, P.; Lászlófi, I.; Hortobábyi, I.; Havasi, G.; Kardos, Z. Preparation of Latanoprostene Bunod of Desired, Pre-Defined Quality by Gravity Chromatography. Patent WO 2017093771 A1, 8 June 2017. [Google Scholar]
- Hortobágyi, I.; Lászlófi, I.; Kardos, Z.; Molnár, J.; Takács, L.; Bán, T. Process for the Preparation of Triple-Bond Containing Optically Active Carboxylic Acids, Carboxylate Salts and Carboxylic Acid Derivatives. Patent WO 2017152667 A1, 14 September 2017. [Google Scholar]
- Hortobágyi, I.; Lászlófi, I.; Kardos, Z.; Molnár, J.; Takács, L.; Tormási, R. Preparation of Epoprostenol Sodium of Enhanced Stability. Patent WO 2017162668 A1, 28 September 2017. [Google Scholar]
- Hortobágyi, I.; Lászlófi, I.; Kardos, Z.; Molnár, J.; Takács, L.; Bán, T. Process for the Optically Active Beraprost. Patent WO 2017174439 A1, 12 October 2017. [Google Scholar]
- Hortobágyi, I.; Lászlófi, I.; Kardos, Z.; Molnár, J.; Takács, L.; Horváth, K. Process for the preparation and purification of misoprostol. Patent WO 2019011668 A1, 19 January 2019. [Google Scholar]
- Hortobágyi, I.; Lászlófi, I.; Varga, Z.; Juhász, I.; Ritz, I.; Kardos, Z. Process for the preparation of polymorph form B of treprostinil diethanolamine salt. Patent WO 2019170093 A2, 12 September 2019. [Google Scholar]
- Rozsumberszki, I.; Váradi, C.; Bán, T.; Kardos, Z.; Hortobágyi, I.; Szabó, T. Process for the preparation of iloprost. Patent WO 2019202345 A2, 24 October 2019. [Google Scholar]
- Corey, E.J.; Arnold, Z.; Hutton, J. Total synthesis of prostaglandins E2 and F2a (dl) via a tricarbocylic intermediate. Tetrahedron Lett. 1970, 4, 307–310. [Google Scholar] [CrossRef]
- Tőke, L.; Szabó, G.; Fogassy, E.; Ács, M.; Nagy, L.; Árvai, L. Verfahren zur Trennung von racemischen Alkalimatellsalzen und vom Lacton der cis-2-Hydroxy-cyclopent-4-en-1-yl-essigsaure. Patent CH 647 496 A5, 9 January 1979. [Google Scholar]
- Sigma-Aldrich Products, Chemical and Biochemical Product. Available online: https://www.sigmaaldrich.com/catalog/search?term=Corey+lactone&interface=All&N=0&mode=match%20partialmax&lang=fr®ion=FR&focus=product (accessed on 26 July 2020).
- Anelli, P.L.; Biffi, C.; Montanari, F.; Quici, S. Fast and selective oxidation of primary alcohols to aldehydes or to carboxylic acids and of secondary alcohols to ketones mediated by oxoammonium salts under two-phase conditions. J. Org. Chem. 1987, 52, 2559–2562. [Google Scholar] [CrossRef]
- Wadsworth, W.S.; Emmons, W.D. The utility of phosphonate carbanion in olefin synthesis. J. Am. Chem. Soc. 1961, 83, 1733–1738. [Google Scholar] [CrossRef]
- Horner, L.; Hofmann, H.; Klink, W.; Ertel, H.; Toscano, V.G. PO aktivierte Verbindung als Olefinierungreagentin. Chem. Ber. 1962, 95, 581–601. [Google Scholar] [CrossRef]
- Bruckner, R.; Harmata, M. (Eds.) Organic Mechanisms—Reactions, Stereochemistry and Synthesis; Springer: Berlin/Heidelberg, Germany, 2010; pp. 471–475. [Google Scholar]
- Corey, E.J.; Bakshi, R.K.; Shibata, S. Highly enantioselective borane reduction of ketones catalyzed by oxazaborolidines. Mechanism and synthetic implications. Am. Chem. Soc. 1987, 109, 5551–5553. [Google Scholar] [CrossRef]
- Brose, S.A.; Thuen, B.T.; Golokovo, M.Y. LC/MS/MS method for analysis of E2 series prostaglandins and isoprostanes. J. Lip. Res. 2011, 52, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Overbeke, A.V.; Baeyens, W.; Oda, H.; Aboul-Enein, H.Y. Direct enantiomeric HPLC separation of several 2-aryl-propionic acids, barbituric acids and benzodiazepines on Chiracel OJ-R chiral stationary phase. Chromatographia 1996, 43, 599–606. [Google Scholar] [CrossRef]
- Chiral Technologies, Daicel-Chromatography Market Leader. Available online: https://mz-at.de/downloads/chiral_chiralcel_oj-rh_instruction_manual.pdf (accessed on 26 July 2020).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Apparatus: | Waters HPLC system equipped with a PDA detector and Empower V3 electronic data processing system |
| Column: | Chiracel OJ-RH, 150 × 4.6 mm, 5 µm |
| Column temperature: | 25 or 40 °C (5–40 °C) * |
| Flow rate: | 0.5 mL/min (0.5–1.0 mL) * |
| Injected volume: | 5 µL |
| Concentration of sample: | 0.25 mg/mL |
| Composition of eluent: | see Table 2 |
| Composition of sample solvent: | acetonitrile:methanol:water 30:10:60 |
| Wavelength: | 200, 210 nm |
| Run time: | 20–40 min |
| Run | Composition | ||
|---|---|---|---|
| Acetonitrile | Methanol | Water (pH = 4) | |
| 1 | 30 | 10 | 60 |
| 2 | 25 | 10 | 65 |
| 3 | 24 | 10 | 66 |
| 4 | 23 | 10 | 67 |
| 5 | 20 | 10 | 70 |
| 6 | 20 | 15 | 65 |
| 7 | 15 | 20 | 65 |
| 8 | 15 | 15 | 70 |
| ent-PG | HPLC Area % | Enantiomeric Excess |
|---|---|---|
| ent-PGF2a.TAM (−)-1.TAM | 98.2 | 0.964 |
| ent-PGE2 (+)-2 | 99.9 | 0.998 |
| ent-PGF1α (−)-3 | 99.3 | 0.986 |
| ent-PGE1 (+)-4 | 99.9 | 0.998 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enesei, M.; Takács, L.; Tormási, E.; Tóthné Rácz, A.; Róka, Z.; Kádár, M.; Kardos, Z. Preparation and Chiral HPLC Separation of the Enantiomeric Forms of Natural Prostaglandins. Chemistry 2020, 2, 727-741. https://doi.org/10.3390/chemistry2030047
Enesei M, Takács L, Tormási E, Tóthné Rácz A, Róka Z, Kádár M, Kardos Z. Preparation and Chiral HPLC Separation of the Enantiomeric Forms of Natural Prostaglandins. Chemistry. 2020; 2(3):727-741. https://doi.org/10.3390/chemistry2030047
Chicago/Turabian StyleEnesei, Márton, László Takács, Enikő Tormási, Adrienn Tóthné Rácz, Zita Róka, Mihály Kádár, and Zsuzsanna Kardos. 2020. "Preparation and Chiral HPLC Separation of the Enantiomeric Forms of Natural Prostaglandins" Chemistry 2, no. 3: 727-741. https://doi.org/10.3390/chemistry2030047