
Preliminary Studies on the Electrochemical Conversion of Liquefied Forest Biomass



Abstract
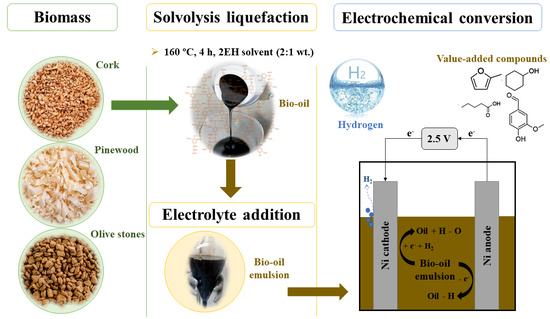
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Bio-Oils Source
2.2. Solvolysis Liquefaction
2.3. Bio-Oils Characterization
2.4. Electrochemical Characterization
2.5. Electrolysis Experiments
3. Results and Discussion
3.1. Solvolysis Liquefaction
3.2. Bio-Oils Characterization
Physicochemical Properties
3.3. Electrochemical Studies
3.3.1. Cyclic Voltammetry Using a Pt Electrode
3.3.2. Linear Scan Voltammetry and Tafel Analysis
3.3.3. Chronoamperometry
3.3.4. Electrolysis Experiments
3.4. Electrochemical Characterization of the Bio-Oil Emulsion Samples after Electrolysis
3.5. Attenuated Total Reflection-Fourier-Transform Infrared Spectroscopy (ATR-FTIR)
- 1750–1735 cm−1, related to the –C=O stretching vibration in esters;
- 1420–1310 cm−1, assigned to –O–H bending vibrations in alcohols;
- 1234–1230 cm−1, the –C–O–C stretching vibration in alkyl aromatics.
- 1420–1310 cm−1, assigned to –O–H bending vibration in alcohols;
- 1205–1020 cm−1, related to –C–O stretching vibration;
- 900–860 cm−1, –C–H bending vibrations.
- 1465–1462 cm−1, related with –C–H deformation;
- 1420–1310 cm−1, –O–H bending vibration;
- 1234–1230 cm−1, –C–O–C stretching vibration.
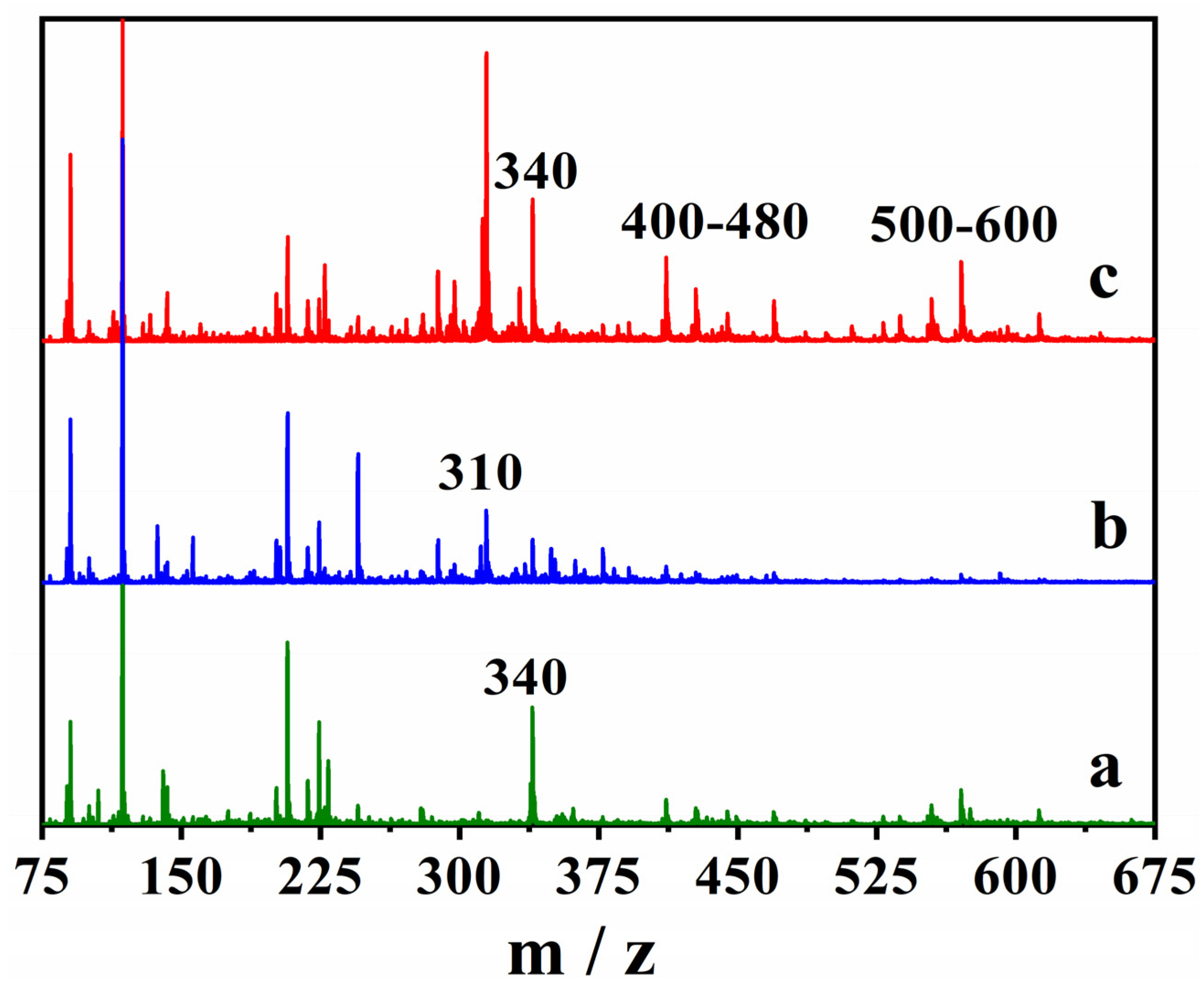
3.6. Mass Spectrometry Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandt, A.; Gräsvik, J.; Hallett, J.P.; Welton, T. Deconstruction of lignocellulosic biomass with ionic liquids. Green Chem. 2013, 15, 550–583. [Google Scholar] [CrossRef] [Green Version]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to biofuels. Green Chem. 2010, 12, 1493–1513. [Google Scholar] [CrossRef]
- Jørgensen, H.; Kristensen, J.B.; Felby, C. Enzymatic conversion of lignocellulose into fermentable sugars: Challenges and opportunities. Biofuels Bioprod. Bioref. 2007, 1, 119–134. [Google Scholar] [CrossRef]
- Wannapeera, J.; Worasuwannarak, N. Upgrading of woody biomass by torrefaction under pressure. J. Anal. Appl. Pyrol. 2012, 96, 173–180. [Google Scholar] [CrossRef]
- Balat, M. Mechanisms of thermochemical biomass conversion processes. Part 3: Reactions of liquefaction. Energ. Source. Part A 2008, 30, 649–659. [Google Scholar] [CrossRef]
- Panwar, N.L.; Kothari, R.; Tyagi, V.V. Thermo chemical conversion of biomass—Eco friendly energy routes. Renew. Sustain. Energy Rev. 2012, 16, 1801–1816. [Google Scholar] [CrossRef]
- Haverly, M.R.; Schulz, T.C.; Whitmer, L.E.; Friend, A.J.; Funkhouser, J.M.; Smith, R.G.; Young, M.K.; Brown, R.C. Continuous solvent liquefaction of biomass in a hydrocarbon solvent. Fuel 2018, 211, 291–300. [Google Scholar] [CrossRef]
- Huang, H.J.; Yuan, X.Z. Recent progress in the direct liquefaction of typical biomass. Prog. Energy Combust. Sci. 2015, 49, 59–80. [Google Scholar] [CrossRef]
- Zhong, C.; Wei, X. A comparative experimental study on the liquefaction of wood. Energy 2004, 29, 1731–1741. [Google Scholar] [CrossRef]
- Behrendt, F.; Neubauer, Y.; Oevermann, M.; Wilmes, B.; Zobel, N. Direct liquefaction of biomass. Chem. Eng. Technol. 2008, 31, 667–677. [Google Scholar] [CrossRef]
- Hu, S.; Wan, C.; Li, Y. Production and characterization of biopolyols and polyurethane foams from crude glycerol based liquefaction of soybean straw. Bioresour. Technol. 2012, 103, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Qin, T.; Huang, L.; Zhang, X.; Yang, Z.; Wang, Y. Mechanisms and main regularities of biomass liquefaction with alcoholic solvents. Energy Fuels 2009, 23, 5213–5218. [Google Scholar] [CrossRef]
- Mateus, M.M.; Carvalho, R.; Bordado, J.C.; dos Santos, R.G. Biomass acid-catalyzed liquefaction—Catalysts performance and polyhydric alcohol influence. Data Brief 2015, 5, 736–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateus, M.M.; Bordado, J.C.; dos Santos, R.G. Potential biofuel from liquefied cork—Higher heating value comparison. Fuel 2016, 174, 114–117. [Google Scholar] [CrossRef]
- Speight, J.G. The Biofuels Handbook; RSC Publishing: Cambridge, UK, 2011. [Google Scholar] [CrossRef] [Green Version]
- Pan, H. Synthesis of polymers from organic solvent liquefied biomass: A review. Renew Sustain. Energy Rev. 2011, 15, 3454–3463. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, J.; Wan, Y.; Lei, H.; Yu, F.; Chen, P.; Lin, X.; Liu, Y.; Ruan, R. Liquefaction of corn stover using industrial biodiesel glycerol. Int. J. Agric. Biol. Eng. 2009, 2, 32–40. [Google Scholar]
- Yu, F.; Liu, Y.; Pan, X.; Lin, X.; Liu, C.; Chen, P.; Ruan, R. Liquefaction of corn stover and preparation of polyester from the liquefied polyol. Appl. Biochem. Biotechnol. 2006, 130, 574–585. [Google Scholar] [CrossRef]
- Lee, S.H.; Teramoto, Y.; Shiraishi, N. Biodegradable polyurethane foam from liquefied waste paper and its thermal stability, biodegradability, and genotoxicity. J. Appl. Polym. Sci. 2002, 83, 1482–1489. [Google Scholar] [CrossRef]
- Dos Santos, R.G.; Carvalho, R.; Silva, E.R.; Bordado, J.C.; Cardoso, A.C.; Costa, M.R.; Mateus, M.M. Natural polymeric water-based adhesive from cork liquefaction. Ind. Crops Prod. 2016, 84, 314–319. [Google Scholar] [CrossRef]
- Esteves, B.; Dulyanska, Y.; Costa, C.; Vicente, J.; Domingos, I.; Pereira, H.; de Lemos, L.T.; Cruz-Lopes, L. Cork liquefaction for Polyurethane foam production. BioResources 2017, 12, 2339–2353. [Google Scholar] [CrossRef]
- Guerra, L.; Moura, K.; Rodrigues, J.; Gomes, J.; Puna, J.; Bordado, J.; Santos, T. Synthesis gas production from water electrolysis, using the Electrocracking concept. J. Environ. Chem. Eng. 2018, 6, 604–609. [Google Scholar] [CrossRef]
- Nilges, P.; dos Santos, T.R.; Harnisch, F.; Schröder, U. Electrochemistry for biofuel generation: Electrochemical conversion of levulinic acid to octane. Energy Environ. Sci. 2012, 5, 5231–5235. [Google Scholar] [CrossRef]
- Dos Santos, T.R.; Nilges, P.; Sauter, W.; Harnisch, F.; Schröder, U. Electrochemistry for the generation of renewable chemicals: Electrochemical conversion of levulinic acid. RSC Adv. 2015, 5, 26634–26643. [Google Scholar] [CrossRef]
- Larew, L.A.; Johnson, D.C. Concentration dependence of the mechanism of glucose oxidation at gold electrodes in alkaline media. J. Electroanal. Chem. 1989, 262, 167–182. [Google Scholar] [CrossRef]
- Adzic, R.R.; Hsiao, M.W.; Yeager, E.B. Electrochemical oxidation of glucose on single crystal gold surfaces. J. Electroanal. Chem. 1989, 260, 475–485. [Google Scholar] [CrossRef]
- Tominaga, M.; Shimazoe, T.; Nagashima, M.; Kusuda, H.; Kubo, A.; Kuwahara, Y.; Taniguchi, I. Electrocatalytic oxidation of glucose at gold-silver alloy, silver and gold nanoparticles in an alkaline solution. J. Electroanal. Chem. 2006, 590, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Aoun, S.B. Electrocatalytic oxidation of glucose at gold nanoparticle-modified pfc electrodes in an alkaline solution. J. Mater. Environ. Sci. 2013, 4, 887–892. [Google Scholar]
- Aoun, S.B.; Taniguchi, I. Effective electrocatalytic oxidation of glucose at platinum nanoparticle-based carbon electrodes. Chem. Lett. 2008, 37, 936–937. [Google Scholar] [CrossRef]
- Parpot, P.; Santos, P.R.B.; Bettencourt, A.P. Electro-oxidation of d-mannose on platinum, gold and nickel electrodes in aqueous medium. J. Electroanal. Chem. 2007, 610, 154–162. [Google Scholar] [CrossRef]
- De Sucre, V.S.; Watkinson, A.P. Anodic oxidation of phenol for waste water treatment. Can. J. Chem. Eng. 1980, 59, 52–59. [Google Scholar] [CrossRef]
- Comninellis, C.; Nerini, A. Anodic oxidation of phenol in the presence of NaCl for wastewater treatment. J. Appl. Electrochem. 1995, 25, 23–28. [Google Scholar] [CrossRef]
- Jokic, A.; Ristic, N.; Jaksic, M.M.; Spasojevic, M.; Krstajic, N. Simultaneous electrolytic production of xylitol and xylonic acid from xylose. J. Appl. Electrochem. 1991, 21, 321–326. [Google Scholar] [CrossRef]
- Governo, A.T.; Proença, L.; Parpot, P.; Lopes, M.I.S.; Fonseca, I.T.E. Electro-oxidation of d-xylose on platinum and gold electrodes in alkaline medium. Electrochim. Acta 2004, 49, 1535–1545. [Google Scholar]
- Weinberg, N.L.; Weinberg, H.R. Electrochemical oxidation of organic compounds. Chem. Rev. 1968, 68, 449–523. [Google Scholar] [CrossRef]
- Andrews, E.M.; Egbert, J.D.; Sanyal, U.; Holladay, J.D.; Weber, R.S. Anode-boosted electrolysis in electrochemical upgrading of bio-oils and in the production of H2. Energy Fuels 2020, 34, 1162–1165. [Google Scholar] [CrossRef]
- Hansen, S.; Mirkouei, A.; Diaz, L.A. A comprehensive state-of-technology review for upgrading bio-oil to renewable or blended hydrocarbon fuels. Renew. Sustain. Energy Rev. 2020, 118, 109548. [Google Scholar] [CrossRef]
- Deng, W.; Xu, K.; Xiong, Z.; Chaiwat, W.; Wang, X.; Su, S.; Hu, S.; Qiu, J.; Wang, Y.; Xiang, J. Evolution of aromatic structures during the low-temperature electrochemical upgrading of bio-oil. Energy Fuels 2019, 33, 11292–11301. [Google Scholar] [CrossRef]
- Lister, T.E.; Diaz, L.A.; Lilga, M.A.; Padmaperuma, A.B.; Lin, Y.; Palakkal, V.M.; Arges, C.G. Low-temperature electrochemical upgrading of bio-oils using polymer electrolyte membranes. Energy Fuels 2018, 32, 5944–5950. [Google Scholar] [CrossRef]
- Dang, Q.; Wright, M.M.; Li, W. Technoeconomic analysis of a hybrid biomass thermochemical and electrochemical conversion system. Energy Technol. 2018, 6, 178–187. [Google Scholar] [CrossRef]
- Silva, T.A.R. Electrochemical conversion of liquefied forest biomass: Preliminary studies. In Thesis to Obtain the Master of Science Degree in Chemical Engineering; Instituto Superior Técnico: Lisboa, Portugal, 2018. [Google Scholar]
- Hariharakrishnan, S. Minimization of fine particles emission from biomass combustion liquefaction, combustion additives and cyclone separators. In Thesis to Obtain the Master of Science Degree in Energy Engineering and Management; Instituto Superior Técnico: Lisboa, Portugal, 2017. [Google Scholar]
- Condeço, J.A.D.; Hariharakrishnan, S.; Ofili, O.M.; Mateus, M.M.; Bordado, J.M.; Correia, J.M.N. Energetic valorisation of agricultural residues by solvent-based liquefaction. Biomass Bioenergy 2021, 147, 106003. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Q.; Lin, X.; Jiang, K.; Han, D. The degradation and repolymerization analysis on solvolysis liquefaction of corn stalk. Polymers 2020, 12, 2337. [Google Scholar] [CrossRef] [PubMed]
- Marques, I.P.; Gil, L.; La Cara, F. Energetic and biochemical valorization of cork boiling wastewater by anaerobic digestion. Biotechnol. Biofuels 2014, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.C.P.; Mateus, M.M.; Santos, D.M.F. On the oxidation of kraft black liquor for lignin recovery: A voltammetric study. J. Electrochem. Soc. 2019, 166, E547–E553. [Google Scholar] [CrossRef]
- Zou, S.; Wu, Y.; Yang, M.; Li, C.; Tong, J. Bio-oil production from sub- and supercritical water liquefaction of microalgae Dunaliella tertiolecta and related properties. Energy Environ. Sci. 2010, 3, 1073–1078. [Google Scholar] [CrossRef]
- Soares, B.; Gama, N.; Freire, C.; Barros-Timmons, A.; Brandão, I.; Silva, R.; Neto, C.P.; Ferreira, A. Ecopolyol production from industrial cork powder via acid liquefaction using polyhydric alcohols. ACS Sustain. Chem. Eng. 2014, 2, 846–854. [Google Scholar] [CrossRef]
- Kanaujia, P.K.; Sharma, Y.K.; Garg, M.O.; Tripathi, D.; Singh, R. Review of analytical strategies in the production and upgrading of bio-oils derived from lignocellulosic biomass. J. Anal. Appl. Pyrol. 2014, 105, 55–74. [Google Scholar] [CrossRef]
- Xu, F.; Yu, J.; Tesso, T.; Dowell, F.; Wang, D. Qualitative and quantitative analysis of lignocellulosic biomass using infrared techniques: A mini-review. Appl. Energy 2013, 104, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Asadieraghi, M.; Daud, W.M.A.W. Characterization of lignocellulosic biomass thermal degradation and physiochemical structure: Effects of demineralization by diverse acid solutions. Energy Convers. Manag. 2014, 82, 71–80. [Google Scholar] [CrossRef]
- Grilc, M.; Likozar, B.; Levec, J. Hydrotreatment of solvolytically liquefied lignocellulosic biomass over NiMo/Al2O3 catalyst: Reaction mechanism, hydrodeoxygenation kinetics and mass transfer model based on FTIR. Biomass Bioenergy 2014, 63, 300–312. [Google Scholar] [CrossRef]
- Akhade, S.A.; Singh, N.; Gutiérrez, O.Y.; Lopez-Ruiz, J.; Wang, H.; Holladay, J.D.; Liu, Y.; Karkamkar, A.; Weber, R.S.; Padmaperuma, A.B.; et al. Electrocatalytic hydrogenation of biomass-derived organics: A review. Chem. Rev. 2020, 120, 11370–11419. [Google Scholar] [CrossRef]
- Carneiro, J.; Nikolla, E. Electrochemical conversion of biomass-based oxygenated compounds. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 85–104. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.H.; Deng, W.; Lang, L.; Jin, X.; Hu, X.; Wang, Y. Minireview on bio-oil upgrading via electrocatalytic hydrogenation: Connecting biofuel production with renewable power. Energy Fuels 2020, 34, 7915–7928. [Google Scholar] [CrossRef]
- Sanyal, U.; Koh, K.; Meyer, L.C.; Karkamkar, A.; Gutierrez, O.Y. Simultaneous electrocatalytic hydrogenation of aldehydes and phenol over carbon-supported metals. J. Appl. Electrochem. 2021, 51, 27–36. [Google Scholar] [CrossRef]
- Qiu, Y.; Lopez-Ruiz, J.A.; Sanyal, U.; Andrews, E.; Gutierrez, O.Y.; Holladay, J.D. Anodic electrocatalytic conversion of carboxylic acids on thin films of RuO2, IrO2, and Pt. Appl. Catal. B 2020, 277, 119277. [Google Scholar] [CrossRef]
- Fereidooni, L.; Tahvildari, K.; Mehrpooya, M. Trans-esterification of waste cooking oil with methanol by electrolysis process using KOH. Renew. Energy 2018, 116, 183–193. [Google Scholar] [CrossRef]
- Moradi, P.; Saidi, M.; Najafabadi, A.T. Biodiesel production via esterification of oleic acid as a representative of free fatty acid using electrolysis technique as a novel approach: Non-catalytic and catalytic conversion. Process Saf. Environ. 2021, 147, 684–692. [Google Scholar] [CrossRef]
- Yuan, G.; Zhang, J.; Zeng, G.; Niu, X.; Wang, L.; Zhang, X.; Wang, Q. Electrocatalytic methyl esterification of fatty acid using boron-doped diamond electrodes. Algal. Res. 2020, 46, 101816. [Google Scholar] [CrossRef]
- Manzoli, M.; Menegazzo, F.; Signoretto, M.; Marchese, D. Biomass derived chemicals: Furfural oxidative esterification to methyl-2-furoate over gold catalysts. Catalysts 2016, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Shao, Y.; Sun, J.; Yin, G.; Du, C.; Wang, Y. Electrocatalytic valorisation of biomass derived chemicals. Catal. Sci. Technol. 2018, 8, 3216–3232. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrolysis # | Cell Design | Anolyte | Catholyte |
|---|---|---|---|
| I | Single-compartment | cork bio-oil with 2 M H2SO4 | |
| II | bio-oil acidic emulsion (50:50 vol.% with 2 M H2SO4 aqueous solution) | ||
| III | bio-oil alkaline emulsion (50:50 vol.% with 2 M KOH aqueous solution) | ||
| IV | Two-compartment | bio-oil acidic emulsion | 2 M H2SO4 |
| V | bio-oil acidic emulsion | ||
| VI | bio-oil alkaline emulsion | bio-oil acidic emulsion | |
| Liquefaction # | 1 | 2 | 3 |
|---|---|---|---|
| Biomass/g | 100 | 200 | 300 |
| Solvent/g | 200 | 400 | 600 |
| Catalyst/g | 6 | 12 | 18 |
| Time/h | 4 | 4 | 4 |
| Temperature/°C | 160 | 160 | 160 |
| Solid residue/g | 33 | 64 | 98 |
| Conversion/% | 67 | 68 | 67 |
| Bio-Oils | Density/g cm−3 | Viscosity/P | Conductivity/mS cm−1 |
|---|---|---|---|
| Cork | 0.88 | <0.1 | 0.5 × 10−3 |
| Pinewood | 0.89 | <0.1 | 1.0 × 10−3 |
| Olive stones | 0.93 | 0.1 | 0.8 × 10−3 |
| Cork + 3 M H2SO4 | 1.04 | 1.5 | 5.34 |
| Pinewood + 3 M H2SO4 | 1.05 | 2.6 | 4.80 |
| Olive stones + 3 M H2SO4 | 1.09 | 19.1 | 1.85 |
| Bio-Oils | H2SO4/M | j2V/mA cm−2 | j−2V/mA cm−2 |
|---|---|---|---|
| Cork | 0 | 0.24 × 10−3 | −0.27 × 10−3 |
| 1 | 0.33 | −0.58 | |
| 3 | 1.10 | −2.23 | |
| Pinewood | 0 | 1.09 × 10−3 | −1.45 × 10−3 |
| 1 | 0.52 | −0.79 | |
| 3 | 1.74 | −4.87 | |
| Olive stones | 0 | 0.99 × 10−3 | −1.23 × 10−3 |
| 1 | 0.23 | −0.34 | |
| 3 | 0.64 | −1.15 |
| Bio-Oils | b/V dec−1 | α | j0/mA cm−2 |
|---|---|---|---|
| Cork | 0.59 | 0.10 | 0.14 |
| Pinewood | 0.59 | 0.10 | 0.31 |
| Olive stones | 0.98 | 0.06 | 0.10 |
| Wavenumber/cm−1 | Functional Group |
|---|---|
| 3600–3100 | –O–H stretching vibration [14,17,47,48,49] |
| 2949–2850 | –C–H stretching vibration [14,17,47,48] |
| 1750–1735 | –C=O stretching vibration in esters [14,47,48] |
| 1725–1705 | –C=O stretching vibration in ketones [47] |
| 1648–1638 | –C=C stretching vibration in alkenes [47] |
| 1650–1561 | –C=C stretching vibration in cyclic alkenes [47] |
| 1465–1462 | –C–H deformation [50] |
| 1420–1310 | –O–H bending vibration in alcohols [51] |
| 1275–1200 | –C–O stretching vibration in alkyl aryl ethers [17] |
| 1234–1230 | –C–O–C stretching vibration in alkyl aromatics [51] |
| 1225–1200 | –C–O stretching vibration in ethers [17] |
| 1205–1020 | –C–O stretching vibration [17,48,49] |
| 980–730 | –C=C bending vibration in alkenes [52] |
| 900–860 | –C–H bending vibration [14] |
| 720–680 | –C–H bending vibration [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, T.; Condeço, J.; Santos, D.M.F. Preliminary Studies on the Electrochemical Conversion of Liquefied Forest Biomass. Reactions 2022, 3, 553-575. https://doi.org/10.3390/reactions3040037
Silva T, Condeço J, Santos DMF. Preliminary Studies on the Electrochemical Conversion of Liquefied Forest Biomass. Reactions. 2022; 3(4):553-575. https://doi.org/10.3390/reactions3040037
Chicago/Turabian StyleSilva, Tiago, José Condeço, and Diogo M. F. Santos. 2022. "Preliminary Studies on the Electrochemical Conversion of Liquefied Forest Biomass" Reactions 3, no. 4: 553-575. https://doi.org/10.3390/reactions3040037