
Preparation of Substituted Pyridines via a Coupling of β-Enamine Carbonyls with Rongalite-Application for Synthesis of Terpyridines


Abstract
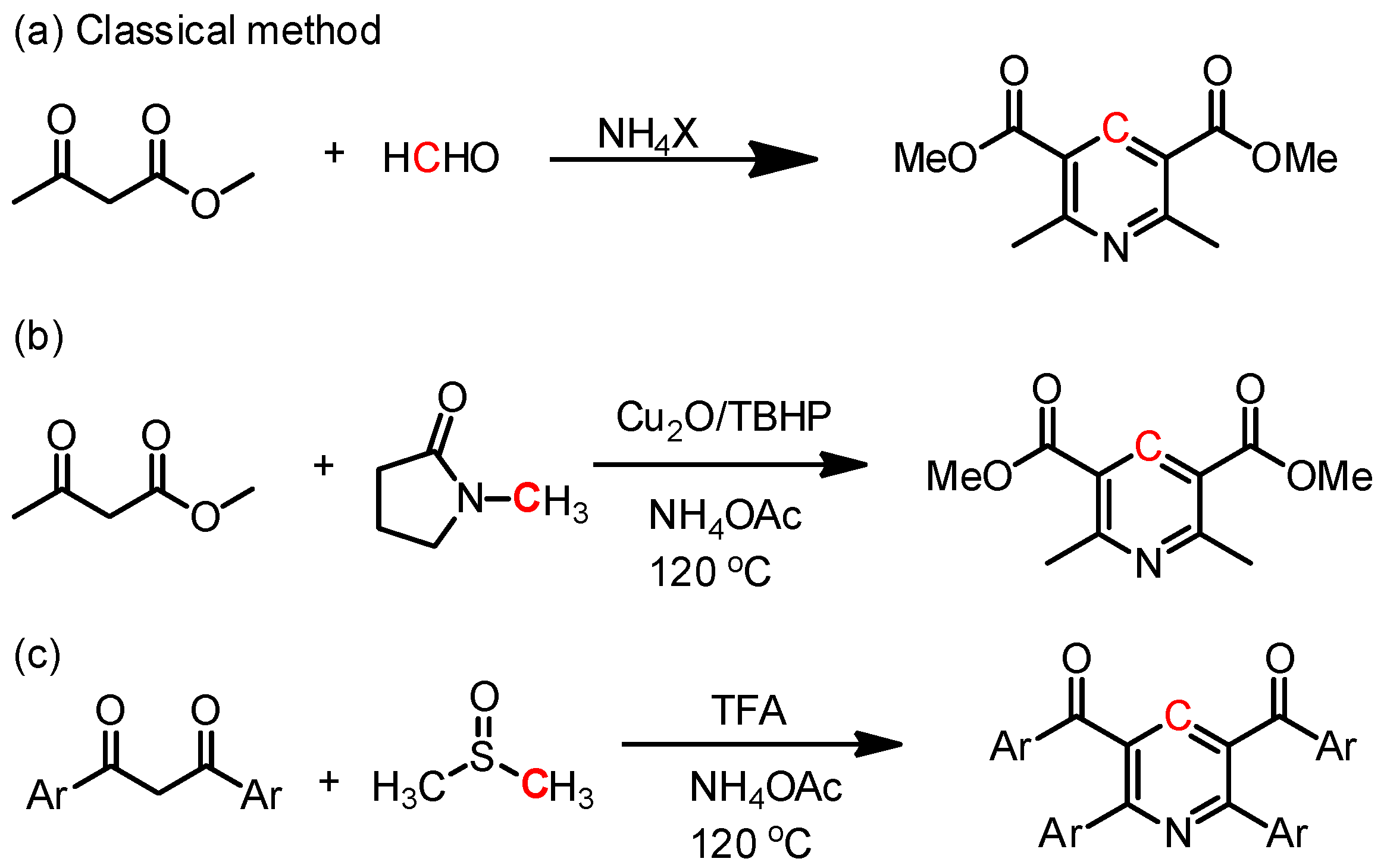
:1. Introduction
2. Materials and Methods
2.1. Materials and Instrumentation
2.2. General Procedure for Preparation of Pyridines and Terpyridines
2.3. Spectroscopic Characterization
3. Results and Discussion
3.1. Optimization of the Reaction
3.2. Reaction Scope
3.3. Synthetic Application-Preparation of Terpyridine
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kleemann, A.; Engel, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances, 5th ed.; Thieme: Stuttgart, Germany, 2000. [Google Scholar]
- Roughley, S.D.; Jordan, A.M. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.X.; Curtis, M.A.; Sperry, J. Pyridine alkaloids with activity in the central nervous system. Bioorg. Med. Chem. 2020, 28, 115820. [Google Scholar] [CrossRef] [PubMed]
- Devadiga, D.; Ahipa, T.N. Recent synthetic advances in pyridine-based thermotropic mesogens. RSC Adv. 2019, 9, 23161–23228. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Iannazzo, D.; Romeo, R.; Giofre, S.V.; Legnani, L. Pyridine and pyrimidine derivatives as privileged scaffolds in biologically active agents. Curr. Med. Chem. 2019, 26, 7166–7195. [Google Scholar] [CrossRef]
- de Ruiter, G.; Lahav, M.; van der Boom, M.E. Pyridine coordination chemistry for molecular assemblies on surfaces. Acc. Chem. Res. 2014, 47, 3407–3416. [Google Scholar] [CrossRef]
- Allais, C.; Grassot, J.-M.; Rodriguez, J.; Constantieux, T. Metal-free multicomponent syntheses of pyridines. Chem. Rev. 2014, 114, 10829–10868. [Google Scholar] [CrossRef]
- Zhou, F.Y.; Jiao, L. Recent developments in transition-metal-free functionalization and derivatization reactions of pyridines. Synlett 2021, 32, 159–178. [Google Scholar]
- Vchislo, N.V. α,β-Unsaturated Aldehydes as C-Building Blocks in the Synthesis of Pyridines, 1,4-Dihydropyridines and 1,2-Dihydropyridines. Asian J. Org. Chem. 2019, 8, 1207–1226. [Google Scholar] [CrossRef]
- Zard, S.Z. The xanthate route to pyridines. Tetrahedron 2020, 76, 130802. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, W.-X.; Xi, Z. Carbodiimide-based synthesis of N-heterocycles: Moving from two classical reactive sites to chemical bond breaking/forming reaction. Chem. Soc. Rev. 2020, 49, 5810–5849. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. Synthesis of heterocycles via palladium-catalyzed carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Shi, M. Divergent synthesis of carbo-and heterocycles via gold-catalyzed reactions. ACS Catal. 2016, 6, 2515–2524. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, K.; Zeng, C. Use of electrochemistry in the synthesis of heterocyclic structures. Chem. Rev. 2018, 118, 4485–4540. [Google Scholar] [CrossRef]
- Hantzsch, A. Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen. Chem. Ber. 1881, 14, 1637–1638. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Conrad, J.; Beifuss, U. Laccase-catalyzed oxidation of Hantzsch 1, 4-dihydropyridines to pyridines and a new one pot synthesis of pyridines. Green Chem. 2012, 14, 2686–2690. [Google Scholar] [CrossRef]
- Yan, Y.; Li, H.; Li, Z.; Niu, B.; Shi, M.; Liu, Y. Copper-catalyzed oxidative coupling of β-keto esters with N-methylamides for the synthesis of symmetrical 2,3,5,6-tetrasubstituted pyridines. J. Org. Chem. 2017, 82, 8628–8633. [Google Scholar] [CrossRef]
- Xue, L.; Cheng, G.; Zhu, R.; Cui, X. Acid-promoted oxidative methylenation of 1, 3-dicarbonyl compounds with DMSO: Application to the three-component synthesis of Hantzsch-type pyridines. RSC Adv. 2017, 7, 44009. [Google Scholar] [CrossRef]
- Ali, R. New dimensions in rongalite chemistry: The land of opportunities in organic synthesis and material sciences. Chem. Select. 2020, 5, 10795–10815. [Google Scholar] [CrossRef]
- Wang, M.; Xiang, J.-C.; Cheng, Y.; Wu, Y.-D.; Wu, A.-X. Synthesis of 2,4,5-trisubstituted furans via a triple C(sp3)–H functionalization reaction using rongalite as the C1 unit. Org. Lett. 2016, 18, 524–527. [Google Scholar] [CrossRef]
- Wang, M.; Tang, B.-C.; Ma, J.-T.; Wang, Z.-X.; Xiang, J.-C.; Wu, Y.-D.; Wang, J.-G.; Wu, A.-X. I2/DMSO-mediated multicomponent reaction of o-hydroxyaryl methyl ketones, rongalite, and DMSO: Access to C3-sulfenylated chromones. Org. Biomol. Chem. 2019, 17, 1535–1541. [Google Scholar] [CrossRef]
- Golla, S.; Anugu, N.; Jalagam, S.; Kokatla, H.P. Rongalite-induced transition-metal and hydride-free reductive aldol reaction: A rapid access to 3, 3′-disubstituted oxindoles and its mechanistic studies. Org. Biomol. Chem. 2022, 20, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-L.; Wu, C.-Y.; Ma, J.-T.; Zhuang, S.-Y.; Yu, Z.-C.; Wu, Y.-D.; Wu, A.-X. Rongalite as C1 synthon and sulfone source: A practical sulfonylmethylation based on the separate-embedding strategy. Org. Lett. 2022, 24, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.M.; Raizada, M.; Sahu, P.K.; Konar, S. Terpyridine-based 3D metal-organic-frameworks: A structure-property correlation. Chem. Eur. J. 2021, 27, 5858–5870. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Schubert, U.S. Metal-terpyridine complexes in catalytic application—A spotlight on the last decade. ChemCatChem 2020, 12, 2841–2890. [Google Scholar] [CrossRef]
- Sakamoto, R.; Wu, K.-H.; Matsuoka, R.; Maeda, H.; Nishihara, H. π-Conjugated bis(terpyridine)metal complex molecular wires. Chem. Soc. Rev. 2015, 44, 7698–7714. [Google Scholar] [CrossRef]
- Housecroft, C.E. 4,2′:6′,4”-Terpyridines: Diverging and diverse building blocks in coordination polymers and metallomacrocycles. Dalton Trans. 2014, 43, 6594–6604. [Google Scholar] [CrossRef]
- Hammarström, L.; Johansson, O. Bis(tridentate) ruthenium–terpyridine complexes featuring microsecond excited-state lifetimes. Coord. Chem. Rev. 2010, 254, 2546–2559. [Google Scholar] [CrossRef]
- Hu, L.; Liu, W.; Li, C.H.; Zhou, X.H.; Zuo, J.L. Iron(II) complexes based on pi-conjugated terpyridine ligands withtetrathiafulvalene or its radical analogue. Eur. J. Inorg. Chem. 2013, 2013, 6037–6048. [Google Scholar] [CrossRef]
- Fang, Y.-Q.; Hanan, G.S. Rapid and efficient synthesis of functionalized bipyridines. Synlett 2003, 2003, 852–854. [Google Scholar] [CrossRef]
- Jameson, D.L.; Guise, L.E. An improved, two-step synthesis of 2,2′:6′,2″-terpyridine. Tetrahedron Lett. 1991, 32, 1999–2002. [Google Scholar] [CrossRef]
- Gade, N.R.; Devendram, V.; Pal, M.; Iqbal, J. IBX mediated reaction of β-enamino esters with allylic alcohols: A one pot metal free domino approach to functionalized pyridines. Chem. Commun. 2013, 49, 7926–7928. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Xu, H.; Zhou, J. Nickel-Catalyzed Asymmetric Transfer Hydrogenation of Olefins for the Synthesis of α- and β-Amino Acids. Angew. Chem. Int. Ed. 2014, 53, 12210–12213. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, M.; Zhao, Y.; Fang, Y.; Zhao, Z.; Xia, B.; Yu, W.; Chang, J. Synthesis of 1,4-Dihydropyridines and Related Heterocycles by Iodine-Mediated Annulation Reactions of N-Cyclopropyl Enamines. Org. Lett. 2021, 23, 9625–9630. [Google Scholar] [CrossRef] [PubMed]
- Anders, P.; Rapp, M.R.; Linseis, M.; Winter, R.F. Tetraruthenium Metallamacrocycles with Potentially Coordinating Appended Functionalities. Inorganics 2018, 6, 73. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Ratio of 1a:2 | Temp | Yield of 3a 2 |
|---|---|---|---|---|
| 1 | MeOH | 1.0:1.0 | 65 °C | 16% |
| 2 | THF | 1.0:1.0 | 65 °C | 13% |
| 3 | EtOH | 1.0:1.0 | 75 °C | 25% |
| 4 | MeCN | 1.0:1.0 | 80 °C | ND |
| 5 | Toluene | 1.0:1.0 | 100 °C | ND |
| 6 | DMF | 1.0:1.0 | 100 °C | 55% |
| 7 | DMSO | 1.0:1.0 | 100 °C | 53% |
| 8 | DMF | 1.0:2.0 | 100 °C | 42% |
| 9 | DMF | 1.0:1.0 | 110 °C | 80% |
| 10 | DMF | 1.0:1.0 | 120 °C | 88% |
| 11 | DMF | 1.0:1.0 | 130 °C | 45% |
| 12 3 | DMF | 1.0:1.0 | 120 °C | 99% |
| 13 3,4 | DMF | 1.0:1.0 | 120 °C | 98% |
| 14 5 | DMF | 1.0:1.0 | 120 °C | ND |
 | ||
|---|---|---|
| Entry | Substituent | Yield 2 |
| 1 | R1 = C6H5 | 3a (95%) |
| 2 | R1 = p-MeC6H4 | 3b (93%) |
| 3 | R1 = p-MeOC6H4 | 3c (95%) |
| 4 | R1 = p-Me2NC6H4 | 3d (84%) |
| 5 | R1 = p-BrC6H4 | 3e (92%) |
| 6 | R1 = p-ClC6H4 | 3f (96%) |
| 7 | R1 = p-IC6H4 | 3g (72%) |
| 8 | R1 = CH3 | 3h (88%) |
| 9 | R1 = furan-2-yl | 3i (83%) |
| 10 | R1 = thiophen-2-yl | 3j (87%) |
 | |||
|---|---|---|---|
| Entry | Substituents of R” | Time | Product (Yield) |
| 1 | –COOMe | 1 h | 9a (78%) |
| 2 | –COCH3 | 20 h | 9b (68%) |
| 3 | –CN | 12 h | 9c (71%) |
| 4 | –COPh | 24 h | 9d (94%) |
| 5 | –CONMe2 | 24 h | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-Y.; Liu, S.-T. Preparation of Substituted Pyridines via a Coupling of β-Enamine Carbonyls with Rongalite-Application for Synthesis of Terpyridines. Reactions 2022, 3, 415-422. https://doi.org/10.3390/reactions3030029
Lee Y-Y, Liu S-T. Preparation of Substituted Pyridines via a Coupling of β-Enamine Carbonyls with Rongalite-Application for Synthesis of Terpyridines. Reactions. 2022; 3(3):415-422. https://doi.org/10.3390/reactions3030029
Chicago/Turabian StyleLee, Yung-Yuan, and Shiuh-Tzung Liu. 2022. "Preparation of Substituted Pyridines via a Coupling of β-Enamine Carbonyls with Rongalite-Application for Synthesis of Terpyridines" Reactions 3, no. 3: 415-422. https://doi.org/10.3390/reactions3030029