Electrocatalysis pivots on the use of electrical energy to apply a potential across a pair of electrodes immersed into a solution of the components to be electrolyzed. In an electrolysis experiment, four key features are present: (1) an anodic oxidation, (2) a cathodic reduction, (3) conservation of the charge in the solution and (4) the presence of a soluble supporting electrolyte to ensure the low electrical resistance of the solution. Common to many organic electrochemical transformations, only one of the two electrodes (working electrode) generates a useful product, while on the counter electrode, a non-productive reaction takes place. For net-oxidative processes, proton reduction to H
2 at the counter electrode is the most common non-productive reaction. For net-reductive reactions, sacrificial oxidation of an amine or the anode itself (e.g., Zn, Mg, Al, Cu) is commonly encountered. In this section, there are many examples in which both electrodes produce useful intermediates. Some common mechanisms in electrolysis are depicted in
Scheme 16 and are those herein presented.
One major difference between the two approaches is that while in photoredox catalysis, the redox events take place on the same site (i.e., the photocatalyst), in electrocatalysis, they occur on two distinct places (cathode and anode). This feature makes it possible to physically separate the two events, when necessary, via the use of a divided cell, provided that a method to accommodate the movement of ions from one half-cell to the other is employed. This is usually achieved by interfacing the two half-cells with a separator, such as a sintered-glass frit, a porous ceramic, a porous polymer sheet or a semipermeable ion-selective membrane (e.g., Nafion). The other fundamental difference is that, in direct electrolysis, redox events occur at the interface between the electrode and solution, which can produce a localized, high concentration of radicals. In contrast, redox chemistry in photoredox catalysis occurs in the solution and provides a low radical concentration. As a result, the coupling of two transient radicals is feasible in electrochemistry if they are generated at the same electrode, whereas this is more challenging in photoredox catalysis.
3.1. Electrocatalyzed Dichlorination of Alkenes
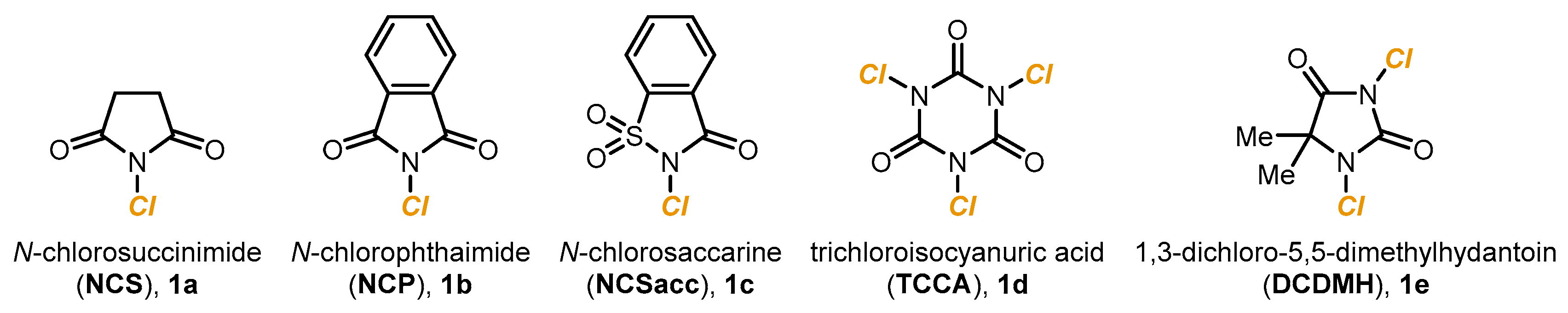
Just like photoredox catalysis is employed (in combination with NCS
1a or chloride salts) to replace Cl
2 in the electrophilic chlorination of arenes, electrocatalysis is a valuable alternative to achieve alkene chlorination under mild and more sustainable reaction conditions. In 2017, Lin described a manganese-catalyzed alkene dichlorination, with MgCl
2 as the chlorine source, producing H
2 and Mg(OAc)
2 as the sole by-products (
Scheme 17) [
34]. Optimizing the dichlorination of indene
55, the authors observed that the direct electrolysis of LiCl on the Pt electrode was not satisfying, giving poor yields of
56 and no
syn:
anti selectivity. Therefore, a redox-active metal was added to impart kinetic control over the difunctionalization, and they obtained good results, in terms of diastereoselectivity, with Mn(OTf)
2. The high diastereoselectivity came from a combination between the steric and electronic properties of the chlorine transfer agent Mn(III)−Cl
57. Further improvements were observed when LiCl was replaced with MgCl
2. The reaction was extended to several styrenes and alkenes, producing the corresponding dichloro derivatives in good yields and diastereoselectivity. Moreover, oxidatively sensitive functional groups (alcohols, aldehydes, amines) were tolerated.
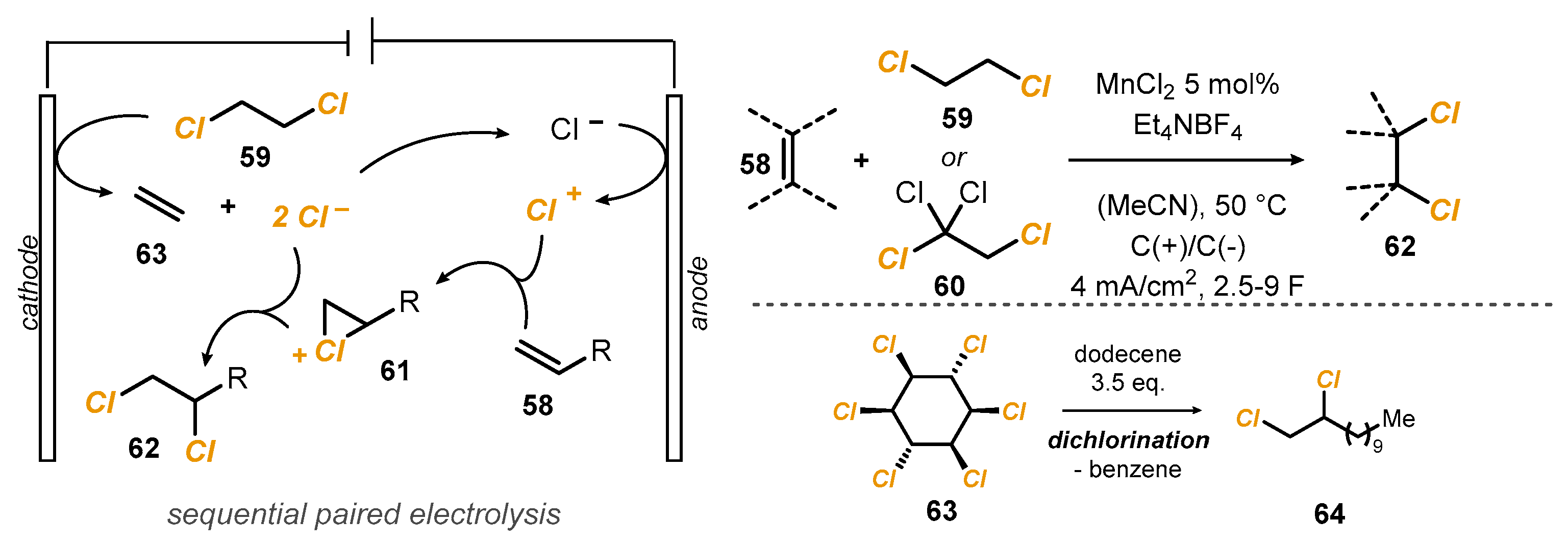
An interesting alternative for the preparation of vicinal dichlorides
62 was recently presented by Morandi. The methodology involved an electronically assisted shuttle paradigm, with inexpensive 1,2-dichloroethane (DCE)
59 used as the chlorine source [
35]. In the cell, two consecutive cathodic reductions of DCE are leveraged to produce chloride anions in the reaction medium, releasing ethene
63. Following this, the oxidation of Cl
− to Cl
+ affords an intermediate chloronium ion
61, which is converted in the final product
62 via ring opening from Cl
− in an overall redox-neutral process (
Scheme 18). Optimal reaction conditions featured graphite electrodes in an Et
4NBF
4 solution (0.1 M in DCE), at 50 °C. MnCl
2 was used as the mediator. In many cases, a solution of 1,1,1,2-tetrachloroethane
60 in MeCN was used instead of neat DCE
59 to obtain better yields. Under optimized conditions, thirty alkenes were efficiently dichlorinated. Noteworthy, soil contaminated with lindane (
gamma-hexachlorocyclohexane)
63 could also be used for the synthesis of 1,2-dichlorododecane
64.
In 2021, Hilt published the first electrochemical
cis-dichlorination of alkenes [
36]. The key reagent in such a transformation was PhSeCl
67, which has a double role (
Scheme 19). On one side, it can be oxidized at the anode, in the presence of tetrabutylammonium chloride (TBAC,
66), to form PhSeCl
3 67a, but on the other side, it can undergo a non-catalyzed
anti-chloroselenation of cyclohexene
68, affording intermediate
67b. PhSeCl
3, being a strong electrophile, may induce a nucleophilic chlorination on intermediate
67b, with inversion of the configuration, producing
cis dichlorocyclohexane
69. The reaction could be applied to cyclic as well as linear olefins, also bearing hydroxyl and acethoxyl moieties. Lower yields were observed on styrenes, and the authors attributed the result to the redox lability of the substrates.
3.2. Electrocatalyzed Heterofunctionalization of Alkenes
After having developed the electrocatalyzed dichlorination of olefins
70, Lin applied an anodically coupled electrolysis to merge two distinct oxidative events and therefore make the heterofunctionalization of alkenes possible. In particular, he developed a protocol to incorporate the trifluoromethyl moiety using Langoi’s reagent
71 (CF
3SO
2Na) [
37]. This salt is directly oxidized on the graphitic carbon to the electrophilic trifluoromethyl radical
71a (anodic event A), capable of reacting with electron-rich alkenes, affording the alkyl radical
71b. Concomitant oxidation of Cl
−, assisted by Mn(OAc)
2 (anodic event B), generates the radical chlorinated reagent Mn(III)-Cl
72, able to quench intermediate
71b, giving the final product
73 (
Scheme 20). The two events could be merged thanks to the different natures of the two radical species. The alkene is indeed more likely to react with a transient free radical, such as
71a, than with the persistent metal-based radical
71c. The authors investigated the scope of the reaction, and several different styrenes and alkenes were efficiently trifluoromethylchlorinated (selected products
74–
76).
The same group later applied a similar concept to achieve alkene chloro-alkylation by anodic generation of a carbon-centered radical from malonitrile through proton-coupled electron transfer [
38].
When anodic and cathodic events are combined, the chloro-chalcogenation of alkenes is possible (
Scheme 21). This was demonstrated by Chen, who reported a cobalt-catalyzed electrochemical oxychlorination of styrenes
77 to α-chloroacetophenones
78 [
39]. To obtain reproducibility, reticulated vitreous carbon (RVC) electrodes were used. Despite prolonging the reaction times, due to the lower current density, they provided better results. Like manganese in the previous examples, cobalt was fundamental in mediating the anodic oxidation of MgCl
2 to a Co(III)-Cl radical species
79a. Oxygen was directly reduced on the cathode to a persistent radical superoxide ion, capable of quenching the transient benzyl radical
79b, affording alcohol
79c. β-Chloroketone
80 was formed by the oxidation of
79c, but the author did not investigate whether the process was electrochemical or not.
With excellent atom economy, Lei used sulfonyl chlorides
81 as bifunctional reagents to produce β-chloro(vinyl)sulfones
83 via a redox-neutral chlorosulfonylation of alkenes and alkynes
82,
Scheme 22 [
40]. In the designed reaction, the cathodic reduction of sulfonyl chloride
81, followed by the cleavage of the S-Cl bonds, produces Cl
− and the sulfur-centered radical
84a, which reacts with the substrate to give intermediate
84b. Chloride anions are not wasted, but oxidized at the anode, with the assistance of MnCl
2, to give Mn(III)-Cl
72. Final radical coupling produces the chlorinated product
83, regenerating the manganese catalyst. The reaction was extended to several styrenes and aryl acetylenes. Moreover, a radical ene–yne cyclization was attempted on propargyl amine
85, and pyrrolidine
86 was isolated in 50% yield.
3.3. Electrocatalyzed Arene Chorination
As highlighted in the Introduction, photocatalysis and electrochemistry are closely related, despite some differences: one among all seems to be the choice of the substrates. Indeed, photoredox catalysis has mostly been used as an alternative mode of activation for aromatic electrophilic substitution, thus affording chloroarenes, with few methodologies applied to alkenes. The opposite trend appears when looking into electrochemistry. The methodology reported thus far in this review is the addition of one of two chlorine atoms to olefins. Nevertheless, there are some recent examples of aromatic chlorination performed under electrochemical conditions.
For example, in 2021, Fang and Guo reported a
C-5 selective chlorination of 8-aminoquinoline
87, catalyzed by copper acetate [
41]. Dichloromethane was used as the source for chlorine radicals, being formed at the anode by the oxidation of small amounts of Cl
− released in the electrolytic cell upon DCM heterolysis, according to the authors’ hypothesis. Aminoquinoline
87 is able to coordinate copper by the two nitrogen atoms, and intermediate
88a can evolve towards product
89 through two different catalytic cycles. One possibility involves the direct addition of the chlorine radical to produce chlorinated intermediate
88b, with the concomitant reduction of the metal center to Cu(I). Anodic oxidation produces arenium ion
88d (
Scheme 23, path B). Alternatively, the same species can be formed from complex
88a after anodic oxidation to radical cation
88c and the subsequent interception of the chlorine radical. In such a catalytic cycle, there is no change in the oxidation state of copper. Wheland intermediate
88c evolves to the final product
89, regenerating the metal catalyst. Protons are reduced at the cathode to equilibrate the overall anodic process. A fairly high current of 100 mA was essential for obtaining satisfying results.
The substrate scope of the reaction was explored, and moderate to excellent yields were obtained when substituted benzamides bearing electron-donating or electron-withdrawing groups were employed. Electron-rich substrates exhibited higher reactivity compared with electron-deficient ones. The different positions of the substituents on the benzene ring did not remarkably affect the reaction efficiency. In addition, alkyl amides were smoothly chlorinated in good yields. The preparation of 89 was also scaled up to the gram scale using a flow electrolytic cell, allowing the synthesis of 1.19 g of product 89 in 24 h (91.2% yield).
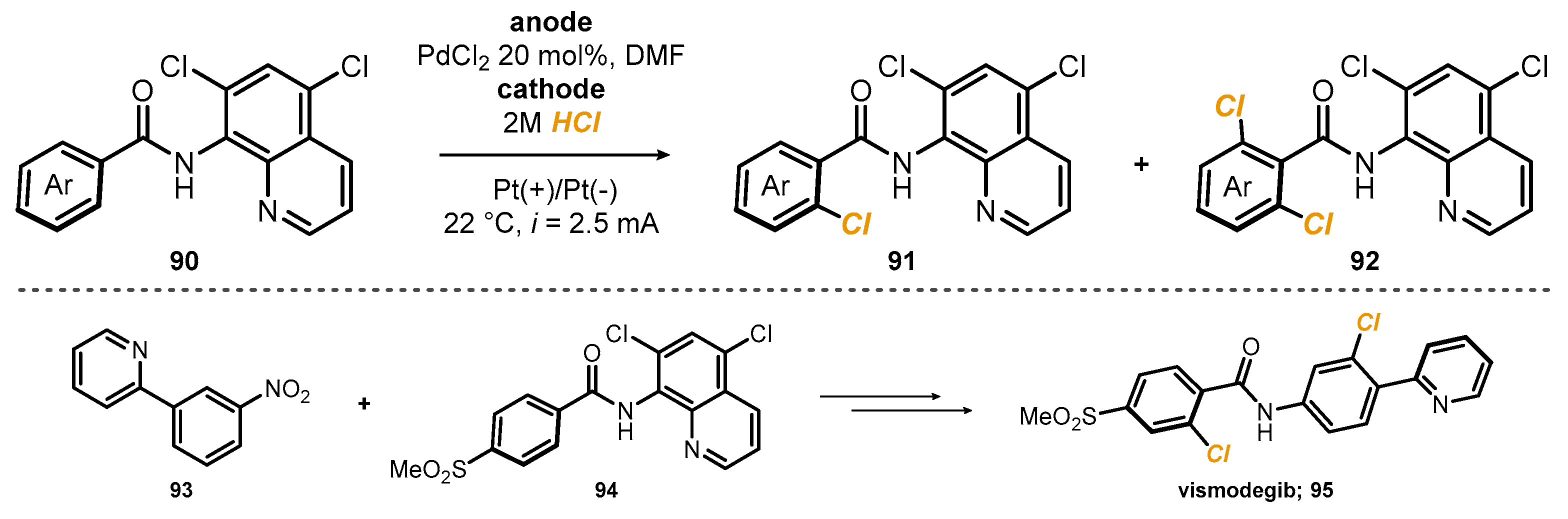
8-Aminoquinolines are peculiar heteroaromatic rings, and due to the distance between the two nitrogen atoms, they have been extensively used as the directing group for the
ortho-functionalization of benzoic acids. The process takes advantage of the proximity between the dicoordinated metal center and the C-H bond, which can undergo metalation, producing a reactive metallacycle. In such a context, palladium is among the most used metals, and an electrochemical version of the process has been reported for the synthesis of
ortho-chlorobenzamides, by Kakiuchi and co-workers [
42]. After the initial screening of the reaction conditions, they observed that unsubstituted 8-aminoquinoline
87 was not the ideal bidentate ligand, since it could undergo chlorination itself, probably by a mechanism similar to the one later exploited by Fang. To avoid such a side reaction, a 5,7-dichloro-8-quinolinyl group was chosen as the directing unit. After testing several conditions, they optimized the process which occurred at 90 °C, with a 100 mA current, in the presence of 10 mol% PdCl
2. Crucial for the success of the reaction was the setup, consisting of a divided cell, which prevented the reduction of palladium at the cathode. Cl
+ or synthetically equivalent species were generated in the anodic semi-cell, with chloride coming from the cathodic reduction of HCl. The reaction was applied to several substituted benzamides
90, and the corresponding chlorinated products
91 were recovered in good to excellent yields (
Scheme 24). In all cases, dichlorination occurred to some extent, producing around 10% of the undesired product
92, with the only exception of
meta-bromo and -CF
3 substituents, which strongly deactivated the aromatic ring toward a second substitution. Finally, palladium-catalyzed C−H chlorination under anodic oxidation conditions was applied to the convergent synthesis of vismodegib
95 from pyridine
93 and benzamide
94.
Prior to the alkene dichlorination using dichloroethane as the source of Cl
+ species reported by Morandi and previously described in this review, Jiao and co-workers explored bifunctional electrocatalysis as a possible way to perform aromatic chlorination, decomposing DCE
59 to vinyl chloride
96 and hydrochloric acid [
43]. They initially investigated the viability of the electrolytic dehydrochlorination of dichloroethane
59, and they observed that a 100 mA current in a solution of
n-Bu
4NOH in DCE
59 led to the formation of vinyl chloride
96 (48%), ethylene
63 (46%) and HCl. Since chloride oxidation to Cl
+ could be coupled at the anode, they tested and demonstrated the possibility of a one-pot aromatic chlorination. Under optimized conditions, they were indeed able to efficiently chlorinate various anilides, bioactive compounds and heterocycles (selected examples
97a–
c) (
Scheme 25).
In the Introduction of this review, we highlighted how the chlorination reaction is among the most straightforward approaches to introduce chloride into molecules, but it comes with several drawbacks when excess chlorine gas is used, such as handling Cl
2 itself and the highly corrosive HCl that is generated. Electrocatalysis could be helpful in this scenario, assuming that chlorine is generated in situ. First, the rate of Cl
2 generation could be regulated by electrochemical parameters, leading to minimum escape from the reaction and facilitating reaction handling under ambient conditions. Second, the presence of an excess of the chloride source could be feasible, avoiding the generation of HCl. In 2021, Cheng et al. published the first example of an electrochemical aromatic chlorination with in situ generated chlorine. The authors identified trichloroacetonitrile
98 as the optimal Cl
2 source, being decomposed on a graphite felt cathode, in the presence of tetraethylammonium chloride as the supporting electrolyte. In optimizing the chlorination of
para-chloroacetamide
99, other solvents were tested (CCl
4, CHCl
3 and DCE), but only traces of product
100 were observed. Moreover, without CCl
3CN, no product was formed, thus excluding a mechanism involving the direct oxidation of the electrolyte. The existence of chlorine was confirmed by the tetrachlorination of
N-tosyl diallylamine, as previously reported by Fu [
34], which excluded a radical pathway. In addition to this, they demonstrated that CCl
3CN
98 could donate all three chlorine atoms, producing acetonitrile. Product
100 was in fact produced in 91% yield after three hours when 0.67 equivalents of CCl
3CN
98 were used. In addition to this, the reaction could also be scaled up, and 153 g (1 mole) of
para-fluoroacetamide
101 gave product
102 in 90% NMR yield and 73% isolated yield (
Scheme 26).
3.4. Electrocatalyzed Miscellaneous Reactions
In the section dedicated to the photochemical functionalization of nitrogen-containing substrates, the C-C bond cleavage on cyclic oxime esters was presented. A similar strategy can be used in electrolysis to access chlorinated products from alcohols, again via ring opening. The concept was presented for the first time by Browne and Morrill, who reported in 2019 the electrochemical deconstructive chlorination of cycloalkanols
103, to synthesize γ- and β-chloroketones
105 [
44]. Additionally, they demonstrated that by employing micro-reactor technology and a recirculating flow, the method could be performed at the gram scale, with incorporated purification. The reaction pivots on the formation of alkoxy radicals directly by O-H homolysis, not a trivial process to achieve under mild reaction conditions, due to the high BDE of RO-H bonds (~105 kcal/mol). The formation of radical
104a does not occur on the electrode surface, but by means of the Mn(III)-Cl species
72, which is also used as an active chlorinating species. The formation of the alkoxy radical also produces HCl, with protons being reduced to hydrogen on the cathode surface. In a similar process to that described by Leonori with oxime esters, alkyl radical
104b is formed upon β-scission from alkoxy radical
104a and, similar to other electrochemical processes, undergoes chlorination by a second equivalent of Mn(III)-Cl
72 (
Scheme 27). The full scope of the electrochemical process was explored starting with the deconstructive chlorination of cyclobutanols. It has been observed that 1-arylcyclobutan-1-ols containing aromatic systems with electron-donating groups at the
ortho or
para positions underwent decomposition. This instability was attributed to the ionization of the C–OH bond in the presence of Brønsted and/or Lewis acids, forming unproductive stabilized carbocations. In such cases, the issue was overcome by employing a syringe pump addition of the substrate over two hours and using tetrabutylammonium acetate as the supporting electrolyte. With a choice of two suitable reaction conditions in hand, they converted a variety of 1-arylcyclobutan-1-ols
103 into the corresponding γ-chlorinated ketones
105 in good to excellent isolated yields. In addition, a representative selection of 1-aryl- and 1-alkylcyclopropan-1-ols
103 could be readily transformed to β-chlorinated ketones
105. Furthermore, to demonstrate scalability, the batch process was translated to a recirculating flow electrochemical setup. Not only did switching from batch to flow allow for a gram-scale synthesis, but also, due to the decreased distance between the electrodes in the flow, the supporting electrolyte was not required, thus making the overall process even more efficient from a sustainability perspective.
Electrochemistry can be exploited not only to achieve synthetically relevant ring-opening processes, but also for the opposite purpose. Indeed, if a nucleophilic group is tethered to the substrate being chlorinated, it is not surprising that a cyclization can be promoted by a careful design of the reaction. One example, from 2020, is the electrochemical dearomative chlorocyclization on tryptamine and tryptophols
106, leading to biologically and pharmaceutically relevant hexahydropyrroloindolines
108 [
45]. Without the help of any metal catalyst, LiCl is oxidized at the anode to an electrophilic Cl
+ species, capable of giving an electrophilic addition to the electron-rich indolic substrate
106, leading to the dearomatized chloronium intermediate
107. The tethered hydroxyl or acetylated amino group is then able, by nucleophilic ring opening, to afford the final tricyclic product
108. Hydrogen evolution at the cathode, from the supporting acetic acid, ensures redox neutrality (
Scheme 28). The method was developed more specifically to obtain brominated derivatives; for such a reason, the scope was restricted to seven chlorinated products. The authors hypothesized the direct anodic oxidation of chloride ions by comparing the yield of chloro (spanning from 50 to 86%) and bromo derivatives (higher than 90%). According to Lei and co-workers, the result was consistent with the higher oxidation potential for Cl
−.
To conclude this section on electrolysis, three examples of non-aromatic C(sp
2)-H chlorination, all from 2021, are reported. The first one, by Liu, is a chlorination of electron-deficient C-H bonds in quinones
109, coumarins
110 1,3-diketones
111 and quinoxalines
112 using six equivalents of HCl [
46]. The reaction occurred under simple experimental conditions, in an undivided cell, with a graphite felt anode and a platinum cathode, using a solution of Et
3NBF
4 in acetonitrile, affording chlorinated products
113–
116 with moderate to good yields (
Scheme 29). Despite the non-aromatic nature of both the starting material and products, the reaction should actually be considered an aromatic chlorination, at least according to the mechanism hypothesized by the authors to explain the chlorination of benzoquinone
109. Cathodic reduction of benzoquinone
109 may easily generate the corresponding aromatic hydroquinone
117, which undergoes electrophilic chlorination with chlorine from the anodic oxidation of Cl
−. Anodic oxidation of the chlorinated hydroquinone
118 gives the desired product. This was not indicated by the authors, but H
2 should be generated at the cathode to ensure redox neutrality.
Liu et al. prepared a series of 3-chlorochromones
120 by the electrochemical chlorination of enaminones
119 [
47]. The reaction was based on a cascade process, initiated by the formation of chloronium intermediate
121a upon the addition of Cl
2 to the electron-rich enaminone group. Chlorine is electrochemically generated on the anode from the oxidation of NaCl. The tethered phenolic moiety opens the chloronium ion by intramolecular formation of a new C-O bond, affording α-chloro emiaminal ether
121b. Elimination of Me
2NH
2Cl produces the final chromone. Additionally, in such a process, protons were reduced on the cathode (
Scheme 30). The scope was restricted to eight examples, but in the same paper, they also applied the reaction to the synthesis of bromo- and iodochromones. The presented method was also scaled up, although a dramatic drop in the yield was already observed on a two-millimole scale.
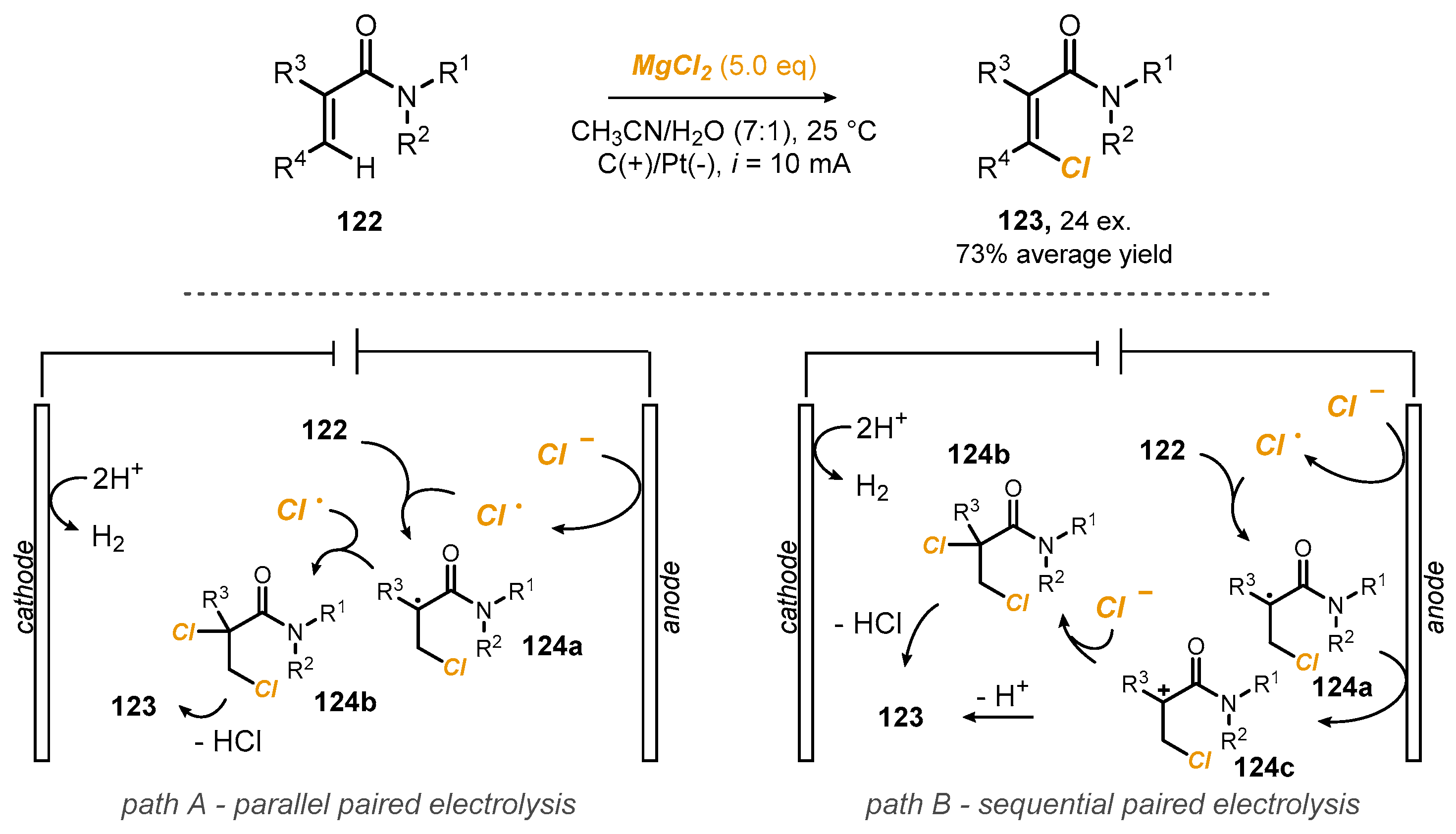
As the last example, Morrill et al. published a method for the oxidative
Z-selective C(sp
2)–H chlorination of tertiary acrylamides
122, providing access to a broad range of synthetically useful
Z-β-chloroacrylamides
123 in good yields,
Scheme 31 [
48]. They optimized a reaction based on MgCl
2 as both the chloride source and electrolyte in MeCN:AcOH (7:1) using galvanostatic conditions (i = 10 mA), a graphite anode and a platinum cathode at 25 °C for 2 h under N
2. More than 20 products were isolated in good to excellent yields (average yield 73%). They observed that several substituents could be incorporated into the C(2)-aromatic unit, including electron-donating, electron-withdrawing and halogen substituents. Additionally, the introduction of sterically demanding groups into the acrylamide starting materials
122 did not negatively impact upon product
123 formation (
Scheme 23). The reaction should initiate with anodic chloride oxidation to form a chlorine radical, which regioselectively adds to the electron-deficient acrylamide
122 to furnish the tertiary C-centered radical
124a. One possibility is then to have a parallel paired electrolysis, with
124a being intercepted by a chlorine radical (path A) to form the dichlorinated intermediate
124b. The final loss of HCl should give acrylamide
122. Alternatively, in a sequential paired electrolysis, radical
124a could be further oxidized to form carbocation
124b, with subsequent deprotonation providing access to the observed product
123, or it could be intercepted by Cl
− to form dichlorinated compound
124c, which could also generate
123 in such a hypothesis via hydrochloric acid loss. In all cases, hydrogen gas should be generated at the cathode.
To demonstrate product utility, the authors succeeded in a palladium-catalyzed Suzuki cross-coupling and in amide hydrolysis. In addition to this, to demonstrate scalability, the batch process was translated to a flow electrochemical setup, employing a syringe pump in combination with a commercially available Ammonite8 flow electro-reactor.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
































