Oxygen Reduction Reaction on Polycrystalline Platinum: On the Activity Enhancing Effect of Polyvinylidene Difluoride


Abstract
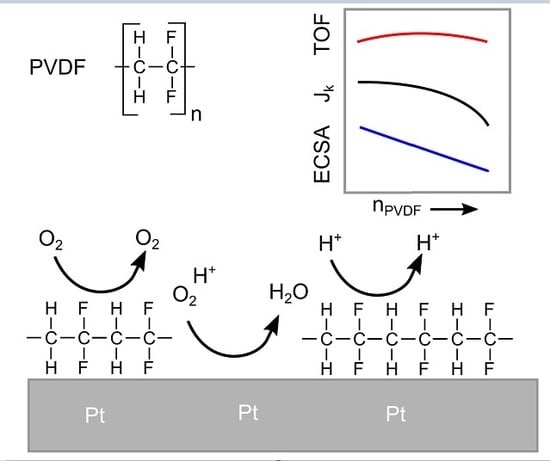
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experiment
2.1. Sample Preparation
2.2. Electrochemical Characterization
3. Results and Discussion
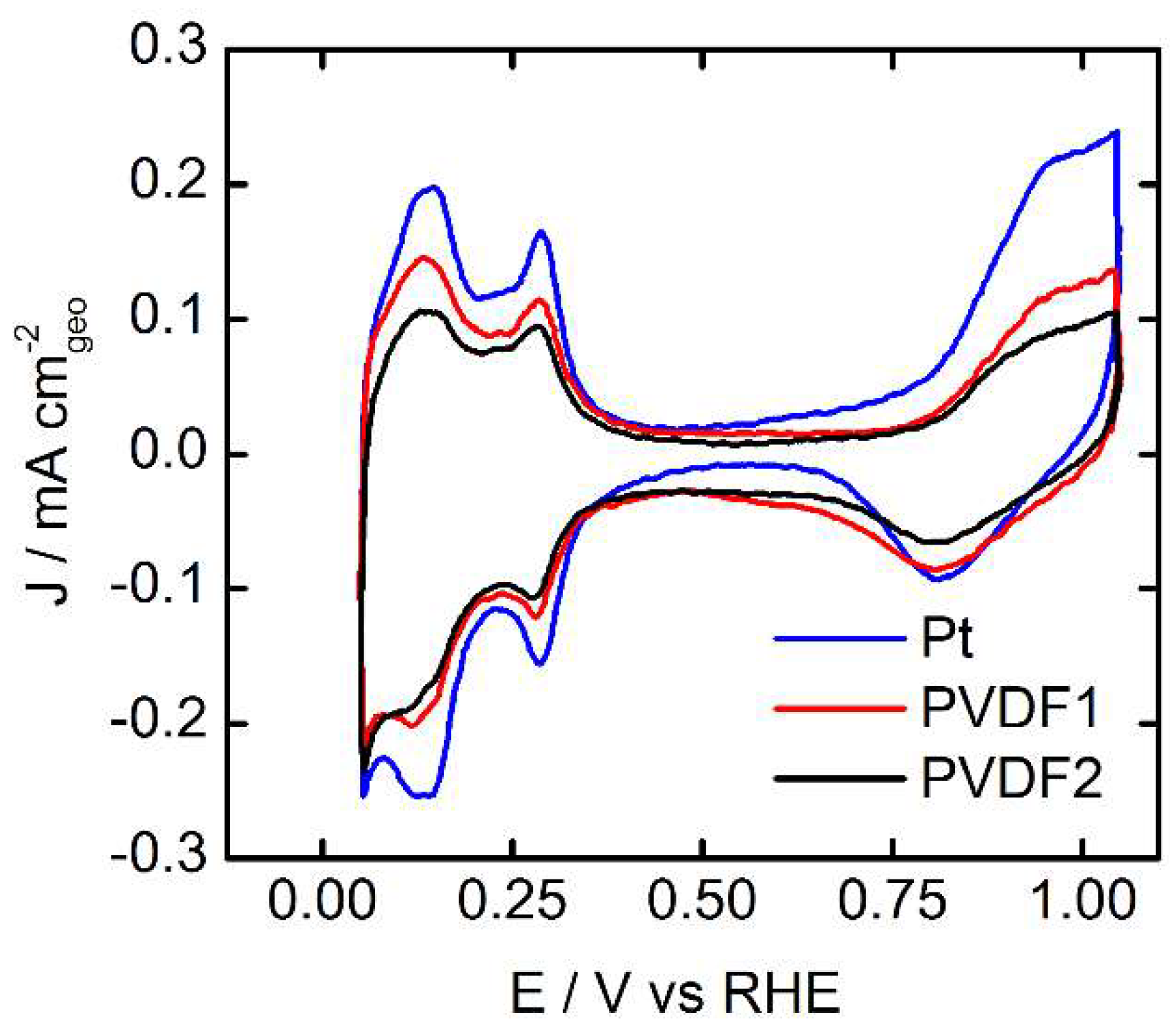
3.1. Surface Characterization in Ar Saturated Electrolyte
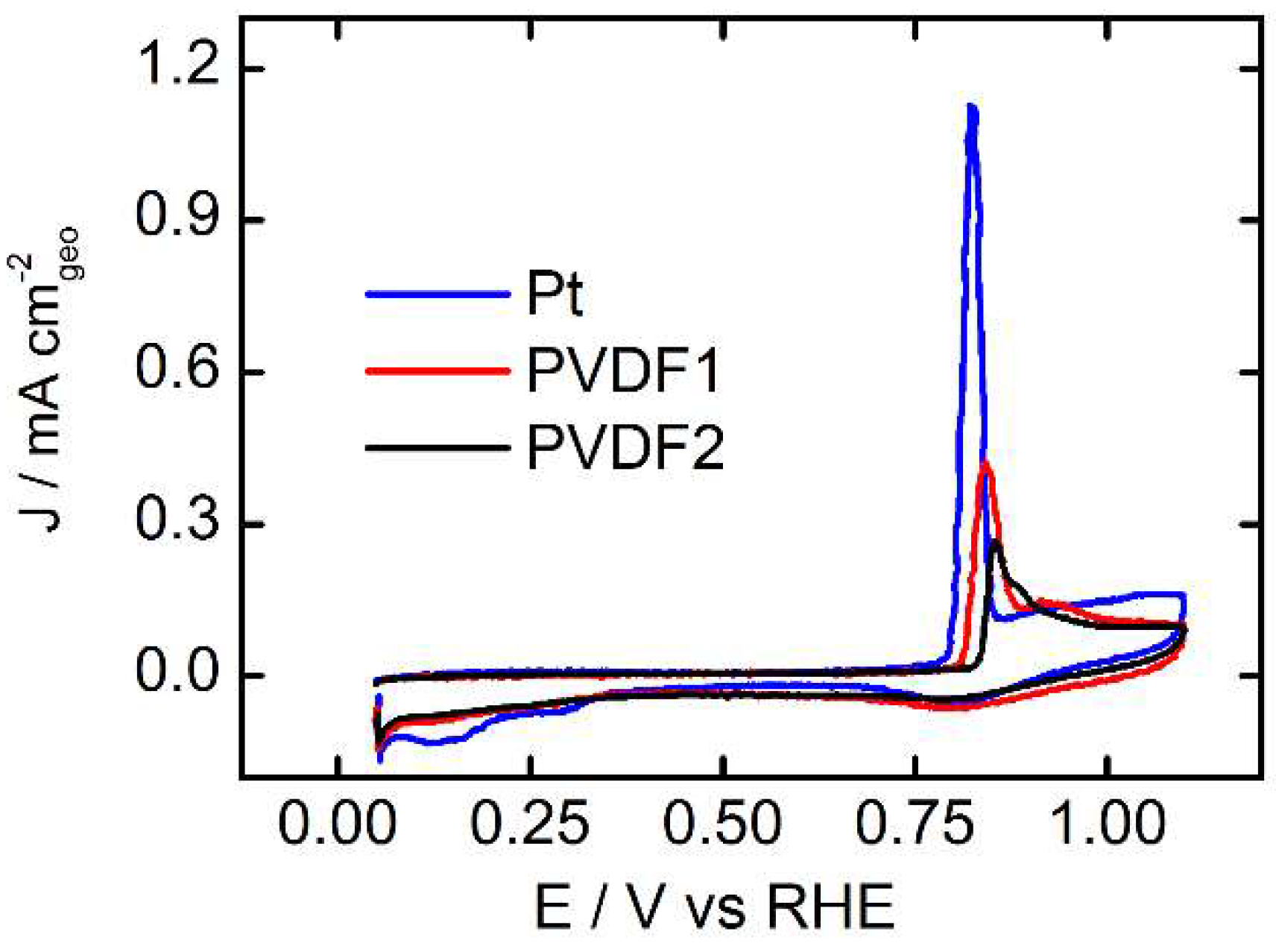
3.2. CO-Stripping Measurements
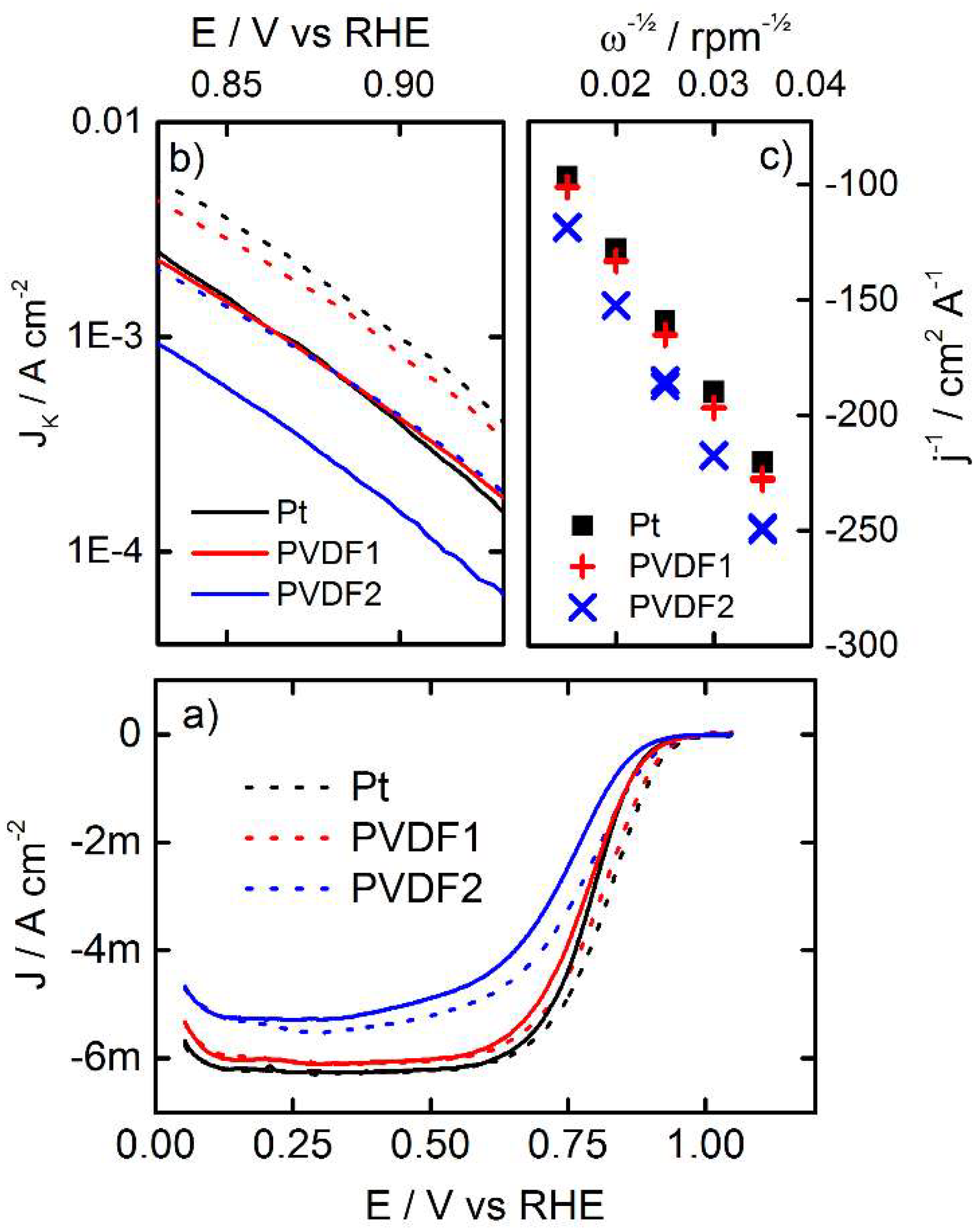
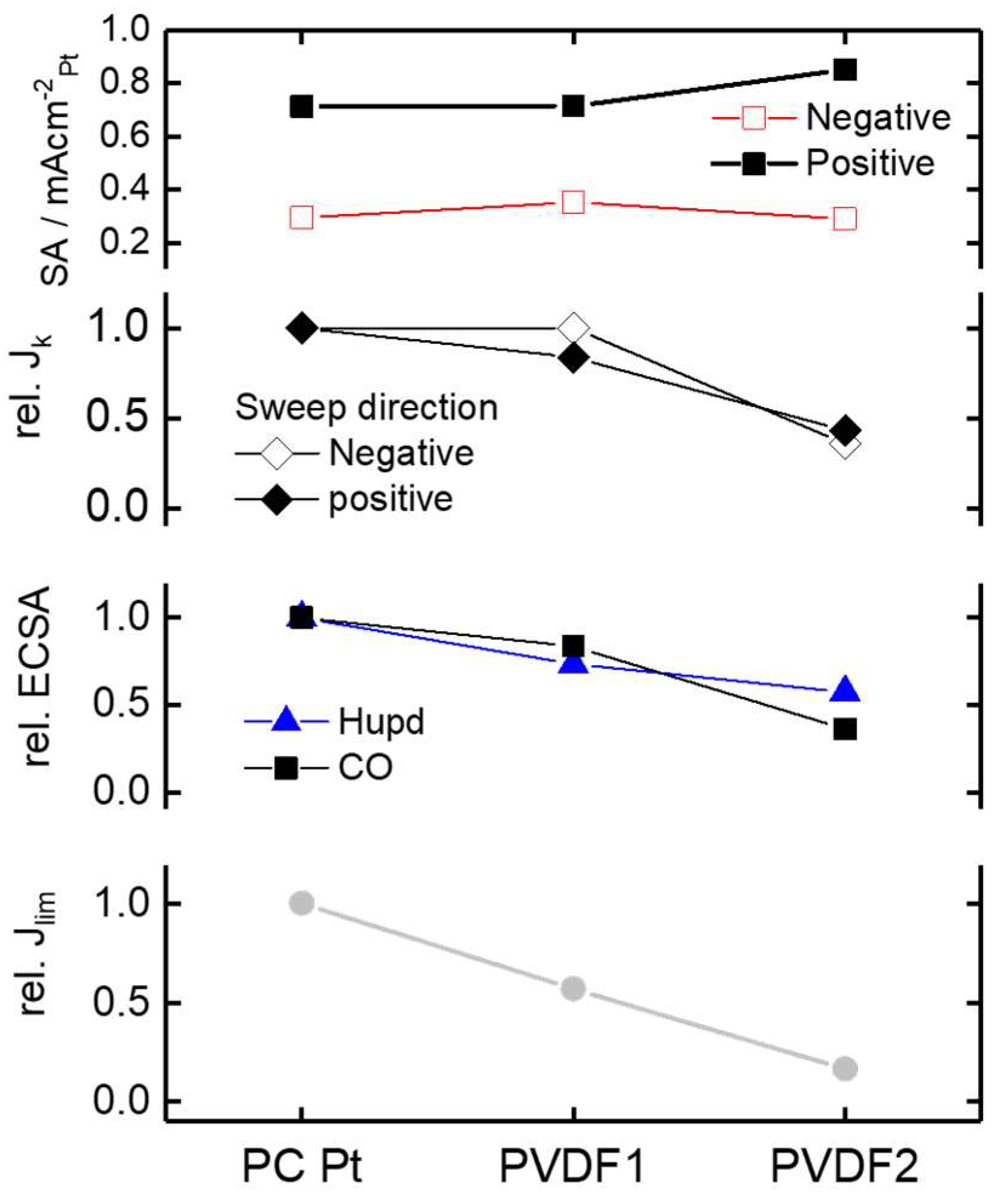
3.3. Oxygen Reduction
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, H.A.; Kocha, S.S.; Sompalli, B.; Wagner, F.T. Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B-Environ. 2005, 56, 9–35. [Google Scholar] [CrossRef]
- Frey, T.; Linardi, M. Effects of membrane electrode assembly preparation on the polymer electrolyte membrane fuel cell performance. Electrochim. Acta 2004, 50, 99–105. [Google Scholar] [CrossRef]
- Shahgaldi, S.; Ghasemi, M.; Wan Daud, W.R.; Yaakob, Z.; Sedighi, M.; Alam, J.; Ismail, A.F. Performance enhancement of microbial fuel cell by PVDF/Nafion nanofibre composite proton exchange membrane. Fuel Process. Technol. 2014, 124, 290–295. [Google Scholar] [CrossRef]
- Li, H.-B.; Shi, W.-Y.; Zhang, Y.-F.; Liu, D.-Q.; Liu, X.-F. Effects of Additives on the Morphology and Performance of PPTA/PVDF in Situ Blend UF Membrane. Polymers 2014, 6, 1846–1861. [Google Scholar] [CrossRef] [Green Version]
- Mu, S.; Tian, M. Optimization of perfluorosulfonic acid ionomer loadings in catalyst layers of proton exchange membrane fuel cells. Electrochim. Acta 2012, 60, 437–442. [Google Scholar] [CrossRef]
- Brodt, M.; Wycisk, R.; Dale, N.; Pintauro, P. Power Output and Durability of Electrospun Fuel Cell Fiber Cathodes with PVDF and Nafion/PVDF Binders. J. Electrochem. Soc. 2016, 163, F401–F410. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.; Gasteiger, H.; Stäb, G.; Urban, P.; Kolb, D.; Behm, R. Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Holst-Olesen, K.; Nesselberger, M.; Perchthaler, M.; Hacker, V.; Arenz, M. Activity inhibition and its mitigation in high temperature proton exchange membrane fuel cells: The role of phosphoric acid, ammonium trifluoromethanesulfonate, and polyvinylidene difluoride. J. Power Sources 2014, 272, 1072–1077. [Google Scholar] [CrossRef]
- Inaba, M.; Quinson, J.; Arenz, M. pH matters: The influence of the catalyst ink on the oxygen reduction activity determined in thin film rotating disk electrode measurements. J. Power Sources 2017, 353, 19–27. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Strmcnik, D.; Blizanac, B.B.; Stamenkovic, V.; Arenz, M.; Markovic, N.M. Measurement of oxygen reduction activities via the rotating disc electrode method: From Pt model surfaces to carbon-supported high surface area catalysts. Electrochim. Acta 2008, 53, 3181–3188. [Google Scholar] [CrossRef]
- Zana, A.; Speder, J.; Roefzaad, M.; Altmann, L.; Bäumer, M.; Arenz, M. Probing Degradation by IL-TEM: The Influence of Stress Test Conditions on the Degradation Mechanism. J. Electrochem. Soc. 2013, 160, F608–F615. [Google Scholar] [CrossRef]
- Nesselberger, M.; Ashton, S.; Meier, J.C.; Katsounaros, I.; Mayrhofer, K.J.J.; Arenz, M. The Particle Size Effect on the Oxygen Reduction Reaction Activity of Pt Catalysts: Influence of Electrolyte and Relation to Single Crystal Models. J. Am. Chem. Soc. 2011, 133, 17428–17433. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, G.K.H. The Development of a State-of-the-Art Experimental Setup Demonstrated by the Investigation of Fuel Cell Reactions in Alkaline Electrolyte. Ph.D. Thesis, Technical Universtity of Munich, Munich, Germany, 2010. [Google Scholar]
- Deng, Y.-J.; Wiberg, G.K.H.; Zana, A.; Arenz, M. On the oxygen reduction reaction in phosphoric acid electrolyte: Evidence of significantly increased inhibition at steady state conditions. Electrochim. Acta 2016, 204, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.-J.; Arenz, M.; Wiberg, G.K. Equilibrium coverage of OH ad in correlation with platinum catalyzed fuel cell reactions in HClO 4. Electrochem. Commun. 2015, 53, 41–44. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O. Real surface area measurements in electrochemistry. Pure Appl. Chem. 1991, 63, 711–734. [Google Scholar] [CrossRef] [Green Version]
- Arenz, M.; Mayrhofer, K.J.J.; Stamenkovic, V.; Blizanac, B.B.; Tomoyuki, T.; Ross, P.N.; Markovic, N.M. The effect of the particle size on the kinetics of CO electrooxidation on high surface area Pt catalysts. J. Am. Chem. Soc. 2005, 127, 6819–6829. [Google Scholar] [CrossRef]
- Wiberg, G.K.H.; Arenz, M. Establishing the potential dependent equilibrium oxide coverage on platinum in alkaline solution and its influence on the oxygen reduction. J. Power Sources 2012, 217, 262–267. [Google Scholar] [CrossRef]
- Hodnik, N.; Baldizzone, C.; Cherevko, S.; Zeradjanin, A.; Mayrhofer, K.J. The Effect of the Voltage Scan Rate on the Determination of the Oxygen Reduction Activity of Pt/C Fuel Cell Catalyst. Electrocatalysis 2015, 6, 1–5. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001; p. 833. [Google Scholar]
- Jinnouchi, R.; Kudo, K.; Kitano, N.; Morimoto, Y. Molecular Dynamics Simulations on O2 Permeation through Nafion Ionomer on Platinum Surface. Electrochim. Acta 2016, 188, 767–776. [Google Scholar] [CrossRef]
- Wiberg, G.K.; Mayrhofer, K.J.; Arenz, M. Investigation of the oxygen reduction activity on silver—A rotating disc electrode study. Fuel Cells 2010, 10, 575–581. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zana, A.; Wiberg, G.K.H.; Arenz, M. Oxygen Reduction Reaction on Polycrystalline Platinum: On the Activity Enhancing Effect of Polyvinylidene Difluoride. Surfaces 2019, 2, 69-77. https://doi.org/10.3390/surfaces2010007
Zana A, Wiberg GKH, Arenz M. Oxygen Reduction Reaction on Polycrystalline Platinum: On the Activity Enhancing Effect of Polyvinylidene Difluoride. Surfaces. 2019; 2(1):69-77. https://doi.org/10.3390/surfaces2010007
Chicago/Turabian StyleZana, Alessandro, Gustav K. H. Wiberg, and Matthias Arenz. 2019. "Oxygen Reduction Reaction on Polycrystalline Platinum: On the Activity Enhancing Effect of Polyvinylidene Difluoride" Surfaces 2, no. 1: 69-77. https://doi.org/10.3390/surfaces2010007