3.1. Pt(111) Electrode
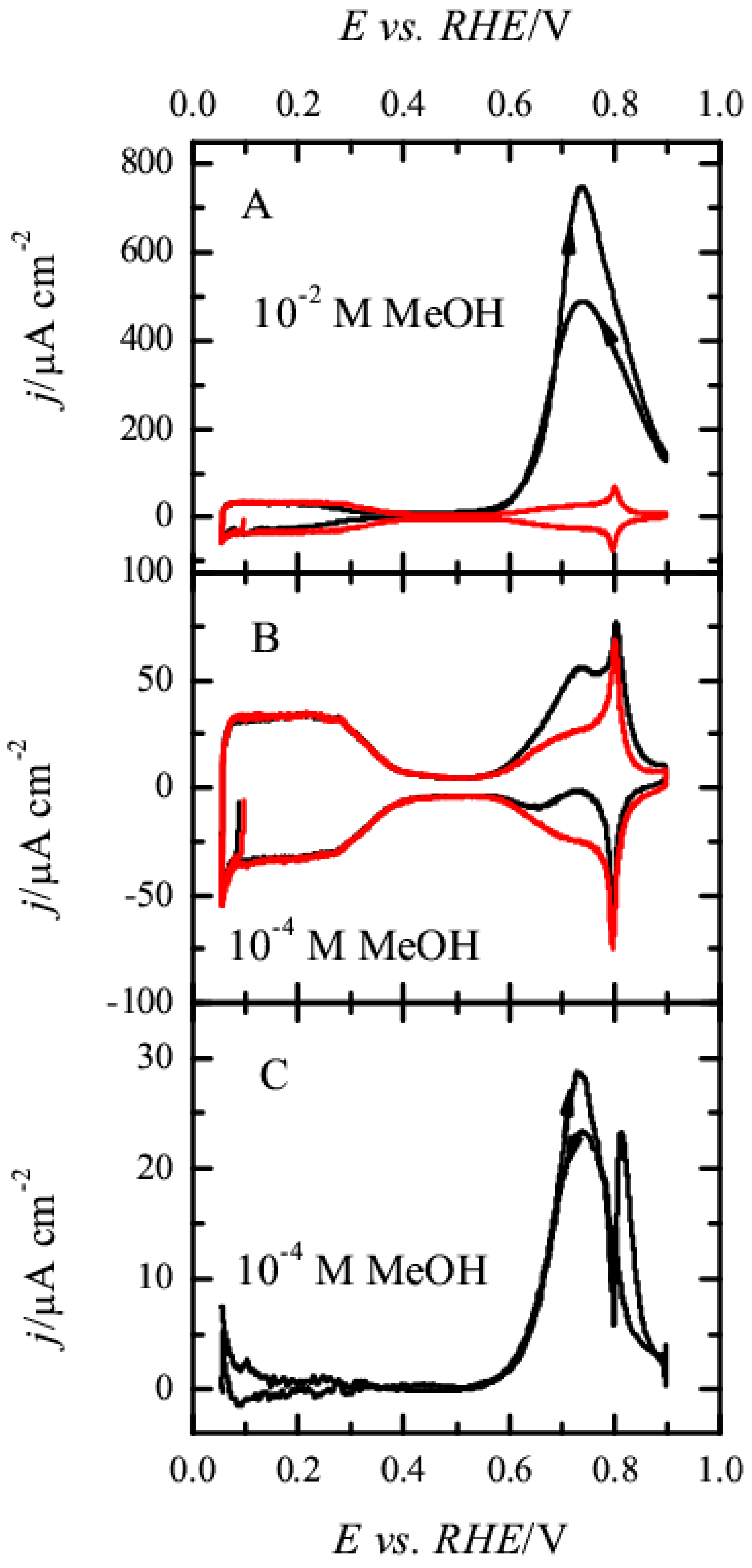
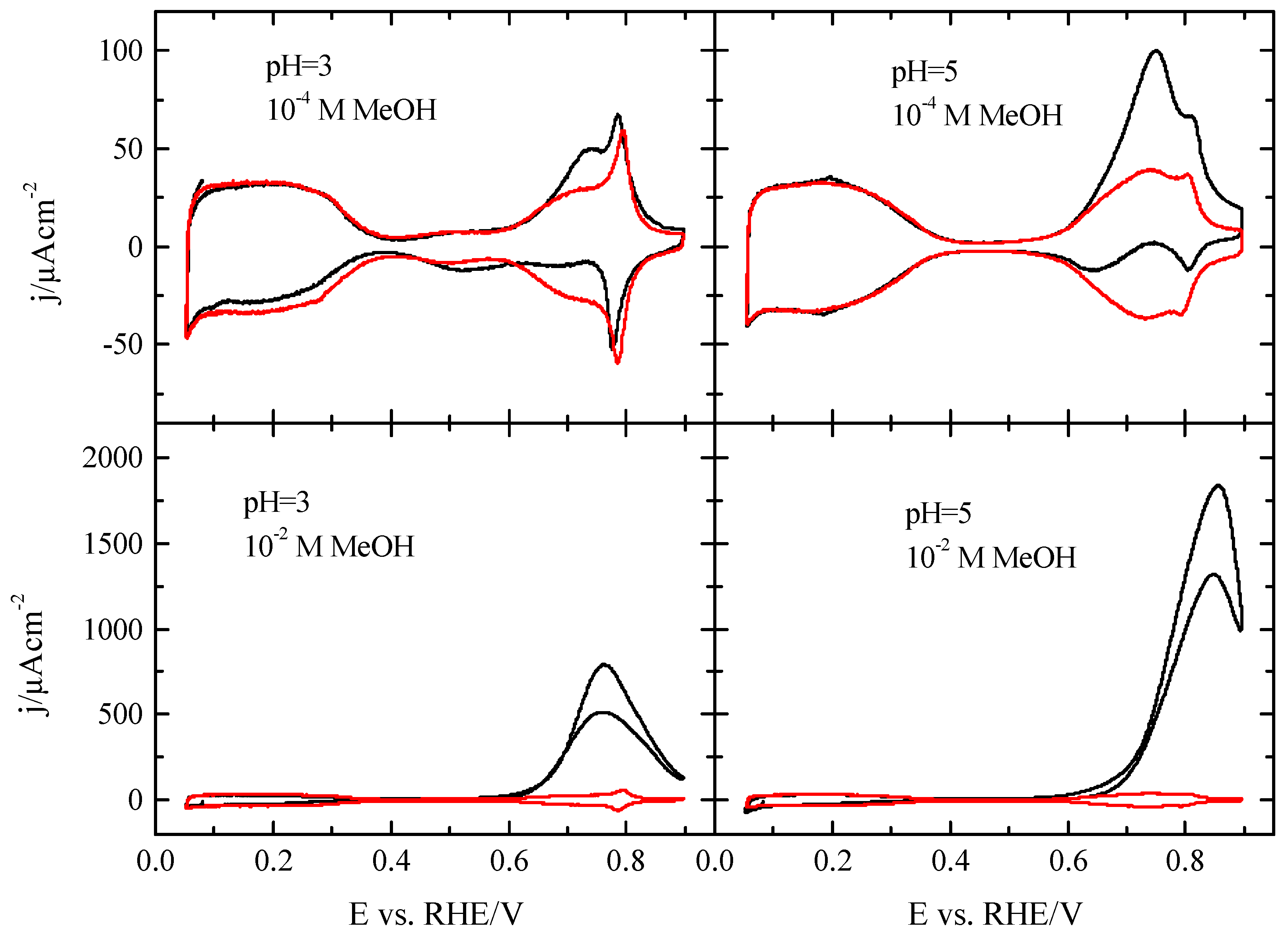
Figure 1 shows the voltammetric profiles of the Pt(111) electrode in 0.1 M HClO
4 with two different methanol concentrations: 10
−2 and 10
−4 M and the comparison with that recorded in the absence of methanol for the same electrolyte solution. The comparison between the blank voltammogram of the Pt(111) electrode and those obtained in the presence of methanol allows correlating the interfacial structure with the reactivity. Moreover, relevant information regarding the oxidation mechanism can be obtained from this comparison, depending on the examined region. As aforementioned, it is generally accepted that methanol oxidation reaction occurs through a dual path mechanism, in which a path involves the formation of adsorbed CO, a poisoning intermediate, and a second path going through an active intermediate. Due to the high number of electrons exchanged in the process, both paths should be interconnected and can share some intermediates leading to a complex mechanism [
6,
7]. The voltammetric profile in the supporting electrolyte can be divided into three regions, hydrogen adsorption region below 0.4 V, the OH adsorption region above 0.6 V, and the double lager region between 0.4 and 0.6 V. The analysis of the modifications of the profiles in these regions will allow obtaining relevant data for the oxidation mechanism.
The profile measured for 10
−2 M has the typical shape of those obtained at higher methanol concentrations, with an effective onset potential of ca. 0.6 V [
14,
24], which coincides with the onset of the OH adsorption process. The large currents measured for this concentration prevents detailed analysis of the effects of the surface composition in the methanol oxidation in this region. To diminish oxidation currents and to establish whether the OH adsorption process is affected by the reaction, currents were recorded for 10
−4 M methanol. For this concentration (
Figure 1B), modifications of the blank voltammetry recorded in the absence of methanol are only observed in the region where OH is adsorbed. Thus, in the positive scan direction, a new peak appears at ca. 0.72 V, apparently superimposed with the original OH adsorption signal, because some of the characteristic features of this region, such as the spike at 0.80 V, are still visible. On the other hand, the profile in the negative scan direction shows complex behavior, which is clearly a consequence of the addition of two different signals: the negative currents associated with the reductive desorption of adsorbed OH and the positive current of the methanol oxidation reaction. To determine whether OH adsorption process is affected by methanol, the blank voltammetric profile obtained in the supporting electrolyte has been subtracted from that measured for the 10
−4 M methanol concentration (
Figure 1C). As can be seen, the resulting voltammetric profile has the same shape as that recorded with larger methanol concentrations, and the only difference is the absolute values of the currents, which are proportional to the methanol concentration. It should be stressed that a spike at 0.80 V in the blank voltammogram, which has been associated to changes in the OH adsorption mode depending on the water structure, is very sensitive to the actual composition of the solution [
25]. The presence of traces of species in concentrations well below 10
−6 M alters the currents in this region. In this case, the subtraction of the two profiles shows a bipolar peak in this region that is only due to the change in the composition of the interface, but not related to the oxidative process of methanol. Then, the subtracted voltammetric profile demonstrates that the OH adsorption process between 0.6 and 0.9 V is also occurring in the presence of methanol and that the interfacial structure of the electrode is not significantly affected by the presence of methanol.
These results clearly show that significant oxidation currents are only recorded in the region where OH is adsorbed, implying that the oxidation of methanol is taking place on a surface partially covered by OH. Moreover, during the spike at 0.80 V, there is a change in the reactivity and the completion of the OH layer at 0.85 V leads to the inhibition of the methanol oxidation reaction. Thus, according to these results, adsorbed OH seems to be required for the oxidation of methanol, but also some free sites where the methanol molecule could interact with the electrode.
The inspection of the hydrogen adsorption and double layer regions can be used to provide information on the type of intermediates that are formed during the oxidation reaction. Probably, CO is the most important intermediate, because its oxidation, which occurs through a well-characterized Langmuir-Hishenlwood mechanism, requires the presence of adsorbed OH on the surface [
26]. Thus, rates for the CO oxidation reaction on Pt are only significant above 0.7 V, and if CO is formed below this potential value, adsorbed CO molecules are accumulated on the surface [
12,
14,
27]. The accumulation of CO on the surface blocks hydrogen adsorption and the hydrogen adsorption charge recorded below 0.4 V diminishes [
28]. As can be seen, in 10
−4 M methanol, the accumulation of CO is negligible because the profile in the hydrogen region for this concentration overlaps with that recorded in the absence of methanol. This fact implies that if CO is formed during the oxidation, it is oxidized at the same rate that is produced and the accumulation during the time of the experiment is negligible. For higher concentrations, there is a small diminution in the hydrogen adsorption charge, implying that some CO has been accumulated on the electrode surface. However, the amount of CO accumulated is very small. In fact, chronoamperometric measurements indicate that CO formation takes place exclusively above 0.5 V and that the rate for this surface is the lowest of all the single crystal electrodes studied [
14].
Additionally, other possible intermediates may be formed and adsorb on the electrode surface giving rise to characteristic signals. One of the possible intermediates in the oxidation of methanol is formic acid. However, the observed behavior is not compatible with the formation of a measurable amount of formic acid. In the acidic media, formic acid adsorbs on the platinum electrode as formate in the bidentate configuration. The adsorption of formate modifies the voltammogram so that the typical shape corresponding to OH adsorption above 0.6 V disappears and a new signal between 0.4 and 0.6 V can be observed [
29]. Moreover, formic acid is readily oxidized on the Pt(111) electrode above 0.35 V [
30,
31]. None of these new signals appear on the recorded voltammograms, even for the highest recorded concentration (0.1 M). Thus, the measured profile between 0.4 and 0.6 V in the presence of methanol matches that recorded in its absence, implying that formic acid is not being formed at significant concentrations during the oxidation of methanol on this electrode surface in the present conditions. It should be mentioned that a small fraction of formic acid has been detected by HPLC analysis after long electrolysis time (ca. 15 min) and with high methanol concentrations [
32]. Clearly, the electrolysis time and the solution concentration affect the product distribution. On the other hand, the presence of methyl formate detected by mass spectroscopy [
13,
27] appears to be a consequence of the attack of a methanol molecule in solution to some intermediate species adsorbed on the surface and not as a result of the esterification reaction between solution methanol and formic acid produced in the partial oxidation of methanol [
33,
34].
From all these results, it can be concluded that the absence of significant modifications in the interfacial structure, in the presence of methanol (aside that relate to the small amount of CO accumulated on the surface), indicates that the different adsorbed intermediates of the reaction are not strongly adsorbed and their interaction with the surface must be weak. In fact, when anions are strongly adsorbed, such as sulfate or phosphate added to the supporting electrolyte, the oxidation currents diminish significantly, because methanol adsorbates cannot compete for the adsorption sites with the strongly adsorbed sulfate or phosphate [
14,
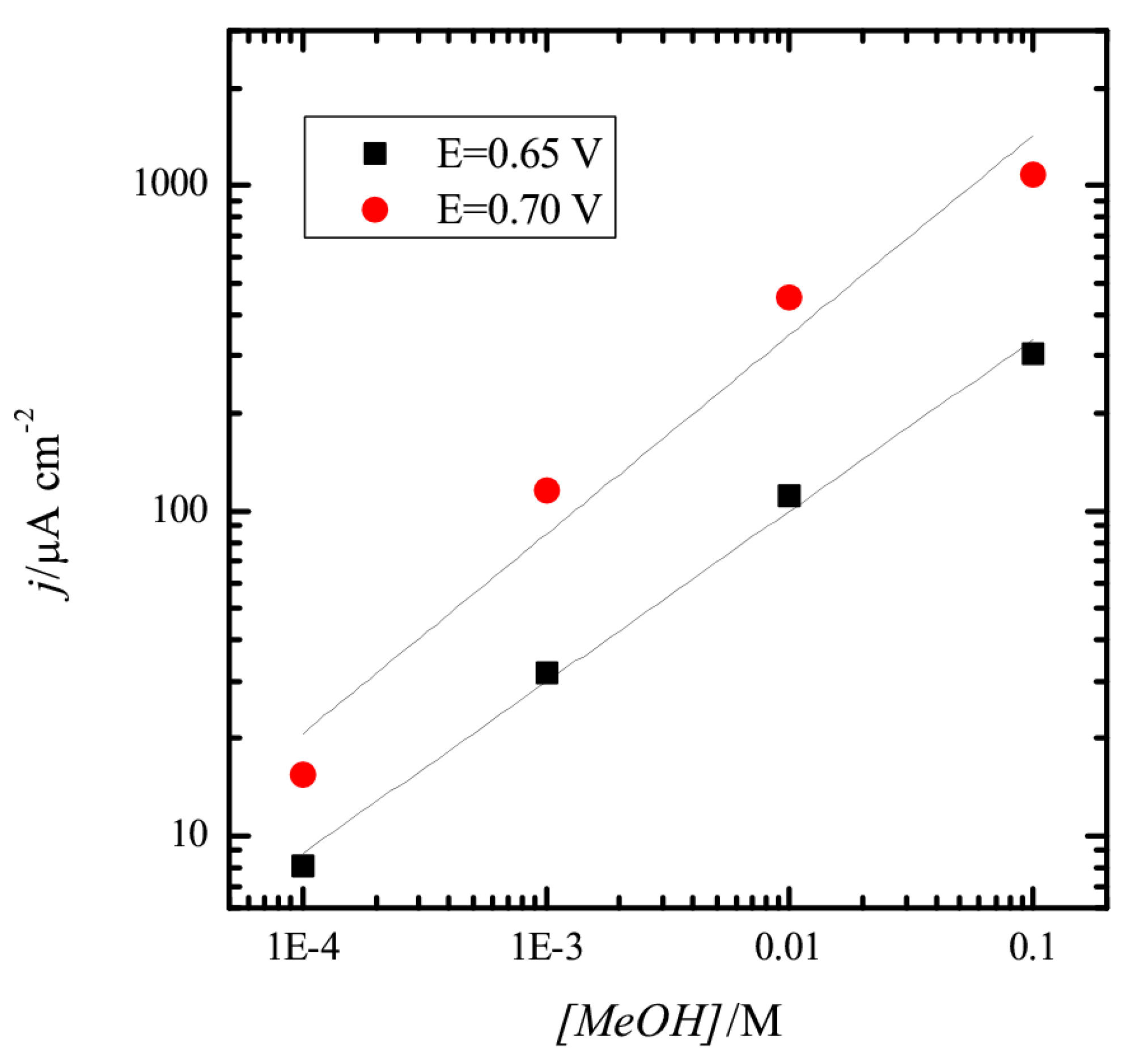
24]. However, the presence of adsorbed OH seems to be required for the oxidation. To further prove this relationship between adsorbed OH and methanol oxidation, the reaction order for methanol was calculated. For this, the current densities measured at 0.65 and 0.75 V after subtraction of the current density at these potentials in the blank voltammogram are plotted vs. the electrode potential in a double logarithmic graph (
Figure 2). As can be seen, a linear relationship is observed for 0.65 V with a slope of 0.53. For 0.70 V, there is a clear deviation from linearity, especially for 0.1 M methanol, where currents are smaller than those predicted from the behavior observed for low concentrations, and the measured slope is 0.62. The fractional reaction orders and the dependence with the electrode potential and OH coverage clearly indicate that the reaction mechanism is complex, and OH should be involved in the process.
Additionally, the comparison with the observed behavior for formic acid will also provide some additional insight. The onset of formic acid oxidation (in the absence of previously formed CO) matches the onset of formate adsorption because the adsorption of formate in the favorable configuration is required for the oxidation [
35,
36]. This adsorption process starts at a potential where hydrogen is desorbed and the adsorption strength of formate is larger than that of OH because OH adsorption is blocked in its presence. On the other hand, the results presented here indicate that the interaction of methanol (and that of the intermediates except for CO) is weak. For this reason, it would be expected that the interaction of methanol with the surface was more favorable in the double layer region because there are no other adsorbed species on the surface. However, the currents in this region are negligible and only where OH is adsorbed, oxidation currents are recorded, reinforcing the relationship between methanol oxidation and adsorbed OH. Additionally, it should be mentioned that the detection of soluble intermediate products increases when sulfuric acid is used as supporting electrolyte [
13,
27,
32]. The presence of adsorbed sulfate prevents the adsorption of OH and the possible intermediates, and thus, the oxidation of methanol becomes more difficult and increasing amounts of partial oxidation products are formed.
Regarding the role of adsorbed OH in this process, previous results with isotopically labeled methanol and water indicate that the cleavage of the C-H bond is involved in the rate determining step [
24]. Then, adsorbed OH should be involved in positioning the methanol molecule in the right configuration for the cleavage and/or in interacting with one of the hydrogen atoms of the methanol molecule in such a way that the activation energy of this (these) step(s) is (are) diminished. This interpretation agrees with the higher amount of partial oxidation products obtained when the adsorption of OH is hindered by the presence of specifically adsorbed anions. This is not the only role of OH in the oxidation mechanism because it is also required for the oxidation of CO in the Langmuir-Hinshelwood mechanism [
26,
37,
38].
Additional effects of the interfacial structure in the oxidation reaction can be obtained by changing the solution pH. Two interfacial parameters are affected, the electrode surface charge and the water structure. The equilibrium potential for the methanol oxidation reaction shifts 59 mV per pH unit, as the RHE scale does. For this reason, it is expected that the methanol oxidation reaction occurs in the same potential region in the RHE scale. However, as the solution pH increases, the electrode potentials for the oxidation in a pH-independent potential scale, such as the Standard Hydrogen Scale (SHE) scale, shift to more negative potential values. Since the potential of zero free charge of the Pt(111) surface is constant and pH-independent (0.28 V vs. SHE [
39]), as the pH increases, the electrode charge becomes more negative, and thus, affecting the interaction of the species with the surface. Additionally, water structure also changes, altering the interaction of the molecules with water, which in turn can influence the reactivity.
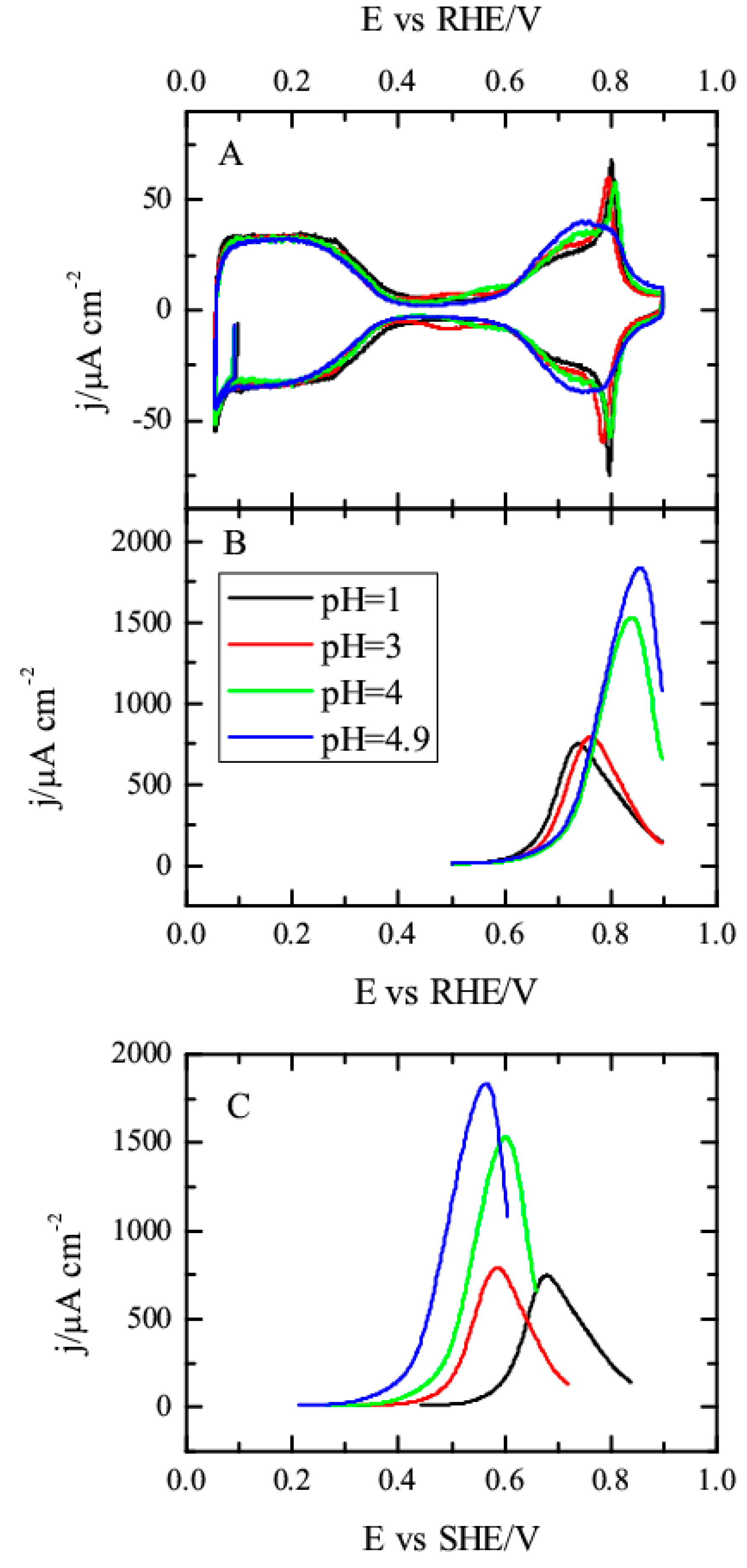
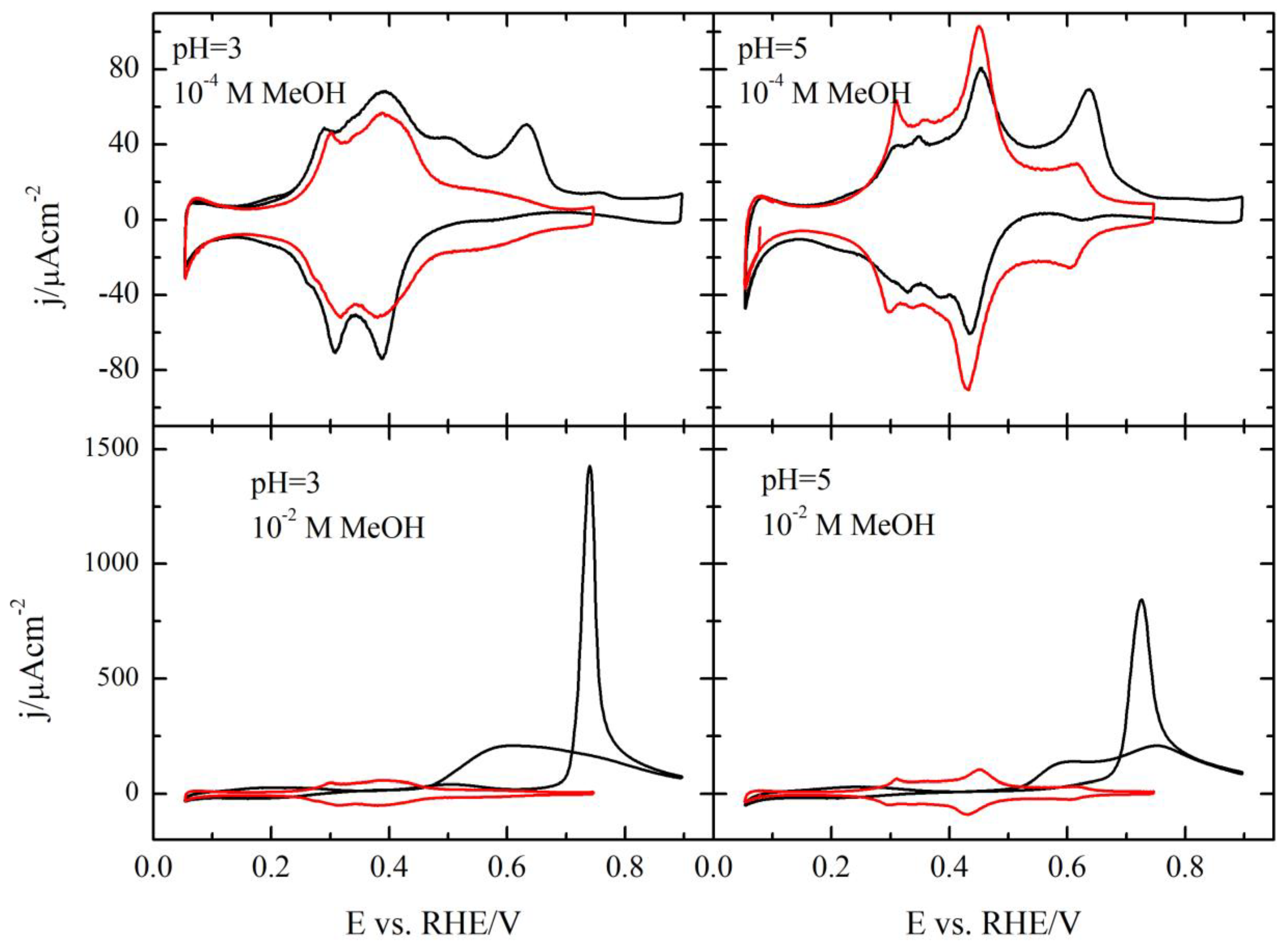
To study those effects, the methanol oxidation reaction was investigated in different pH solutions up to a pH value of 5 (
Figure 3). As can be seen, the qualitative behavior of the electrode is independent of the solution pH, and the oxidation of methanol is only taking place in the region where OH is adsorbed. Thus, at 10
−4 M methanol, an oxidation wave is observed as superimposed with the OH adsorption in the positive scan direction, whereas in the negative direction, the profile is more complex, as happens for pH = 1. Additionally, the profile in the hydrogen region overlaps with the measured in the absence of methanol, which implies that CO is not accumulated on this surface. For higher concentrations, the general shape of the voltammogram in methanol containing solutions do not change with the pH, showing a broad wave, whose onset potential coincides with that of OH adsorption and currents in the negative scan direction, which are slightly smaller than those recorded in the positive direction. However, a detailed analysis shows some modifications with pH, as discussed below.
The detailed analysis of the different effects require that the profiles at different pH values are compared.
Figure 4 shows the voltammetric profiles of the Pt(111) electrode in the presence and absence of methanol for the different pH values. As can be seen, the changes in the blank voltammetric profile are small, especially for pH values lower than four. The onset for the OH adsorption is the same and the only major modifications are observed for the spike, which tends to disappear and as the solution pH increases, in a clear evolution to the profile recorded in alkaline media [
40]. Despite these minimal changes, the oxidation of methanol is clearly affected by the pH. As shown in
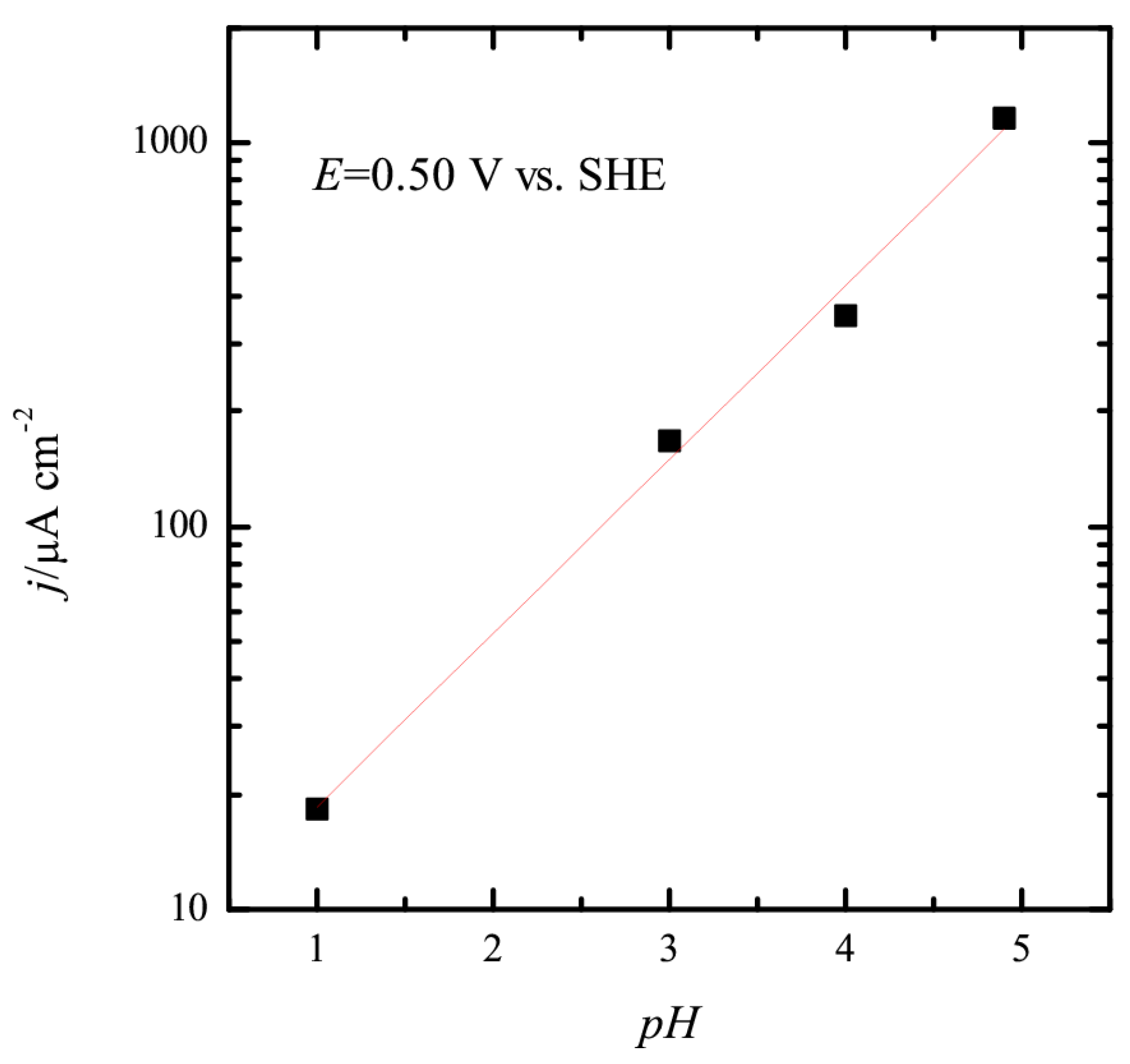
Figure 4B, the onset of the oxidation displaces to more positive potentials in the RHE scale and the current increases. Clearly, the changes in the interfacial properties are affecting this reaction. Additional information on that can be obtained calculating the reaction order for protons. For an irreversible reaction, reaction orders should be calculated at a constant potential value, in this case, in the SHE scale. Thus, when plotting the measured current at 0.5 V vs. SHE as a function of the pH in a double logarithm scale, a linear relationship is obtained in which the slope is ca. 0.45 (
Figure 5). This value implies that the reaction order with respect to the proton concentration is –0.45. While the fractional orders are difficult to explain with simple mechanisms, the straightforward interpretation would be that protons are released before the rate-determining step. This observed behavior is in good agreement with the isotopic effect observed [
11], which also indicate that the release of protons is involved in the rate determining step. An additional effect that should be taken into account when analyzing this behavior is the effect of the surface electrode charge. Since the potential of zero free charge is 0.28 V vs. SHE, as the pH increases, the region in which methanol oxidation takes place moves from positive charge to neutral values. Thus, the interaction between the species involved in the reaction (OH and methanol) and the surface is modified and affects the reaction rates. In this sense, for the oxygen reduction reaction, it has been proposed that higher rates obtained in neutral values are related to the surface charge values [
41]. For neutral pH values, the charge of the surface is neutral in the onset of the ORR. The observed behavior for methanol shows a very similar dependence.
3.2. Pt(100) and Pt(110) Electrodes
The major difference between the behavior of the Pt(111) electrode and the other two basal planes is the presence/absence of a pure double layer region where no specific adsorption occurs. For the Pt(110) and Pt(100) electrodes, OH is readily adsorbed at low potentials when hydrogen is being desorbed [
23,
42,
43,
44,
45], and thus there is no pure double layer region. Then, any molecule that interacts with the surface should either compete with hydrogen or OH for the adsorption sites. Additionally, the overlapping of the two regions prevents the determination of the potential of zero free charge.
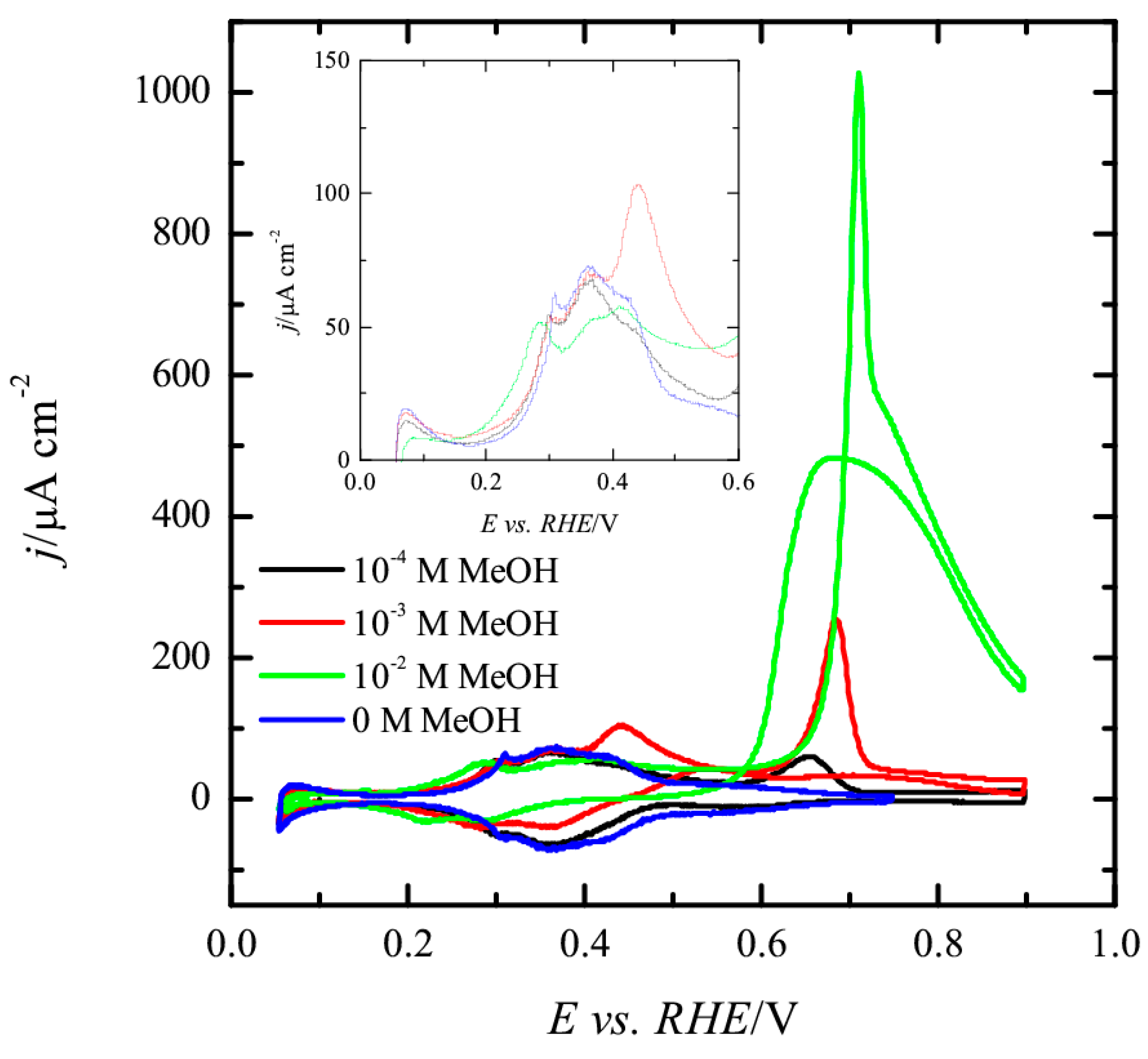
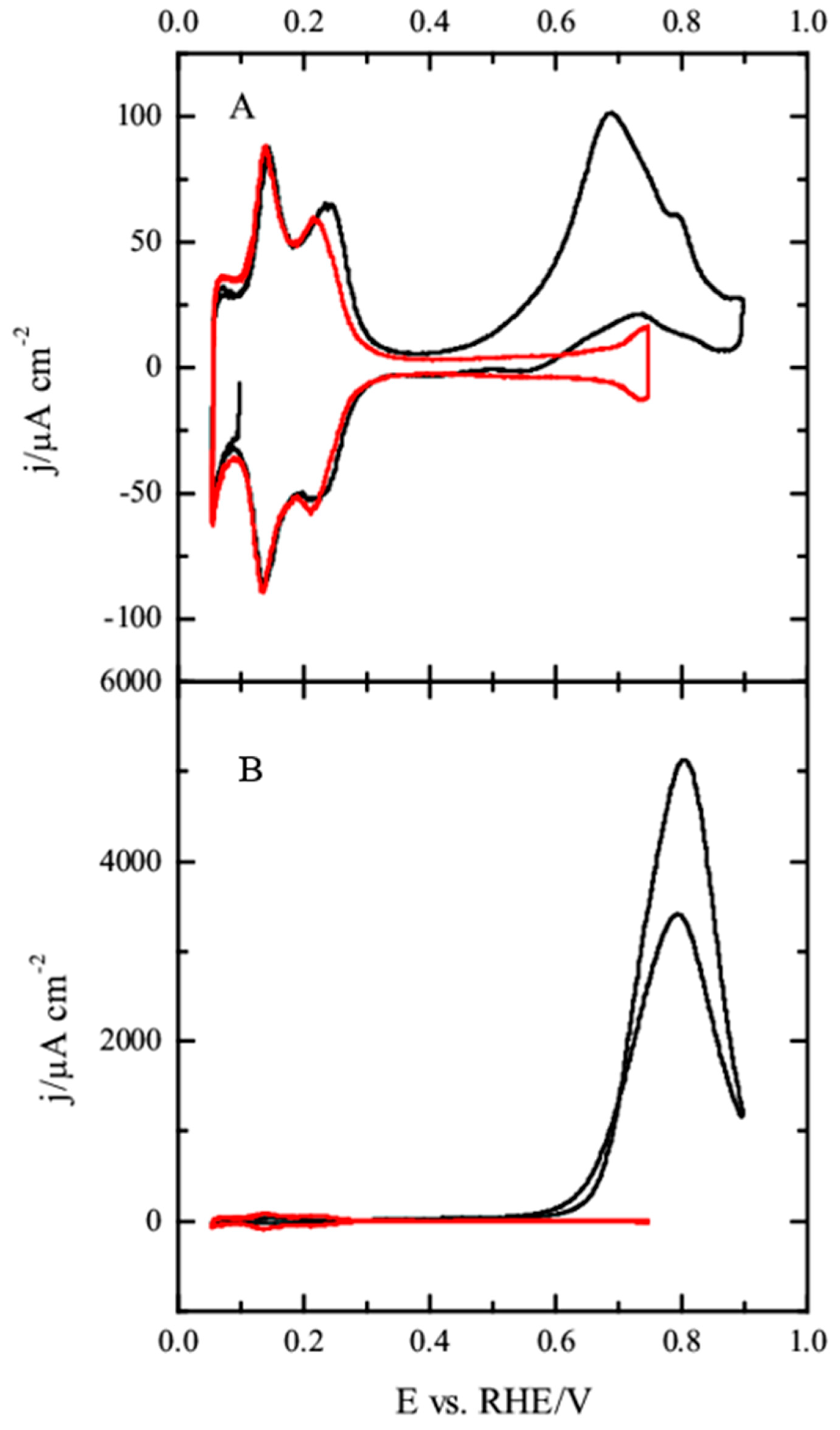
For the Pt(100) electrode (
Figure 6), hydrogen is adsorbed on the surface between 0.05 and 0.45 V and the OH adsorption occurs in a broad wave between 0.45 and 0.7 V, just after the desorption of hydrogen from the surface [
43,
45]. In the presence of 10
−4 M methanol, the only significant modification is the appearance of a peak at ca. 0.65 V in the positive scan direction, and a small diminution in the hydrogen adsorption charge. Unlike the Pt(111) electrode, this peak is only visible in the positive scan direction, and thus should be related to the oxidation of some CO molecules that have been accumulated on the electrode surface at low potentials. However, the amount of CO accumulated on the surface is very small and negligible currents are recorded in the negative scan direction. As the methanol concentration increases, this CO oxidation peak increases in charge and shifts to more positive potentials. In parallel, hydrogen adsorption charge below 0.45 V diminishes due to the blockage of hydrogen adsorption sites by CO. Additionally, a second oxidation wave after this peak CO emerges both in the positive and negative scan directions. This wave shows a hysteresis between both scan directions, which is clearly visible for the solution containing 10
−2 M methanol, and should be related to the accumulation of CO in the positive scan direction at potentials below 0.6 V. Thus, the presence of adsorbed CO in the positive scan direction hinders the oxidation of additional molecules of methanol on the surface. After the oxidation of CO, methanol molecules can interact with the surface and give additional oxidation current contributions. In the negative scan direction, any CO formed during the reaction can be readily oxidized at potentials higher than 0.6 V, giving rise to the hysteresis between both scan directions.
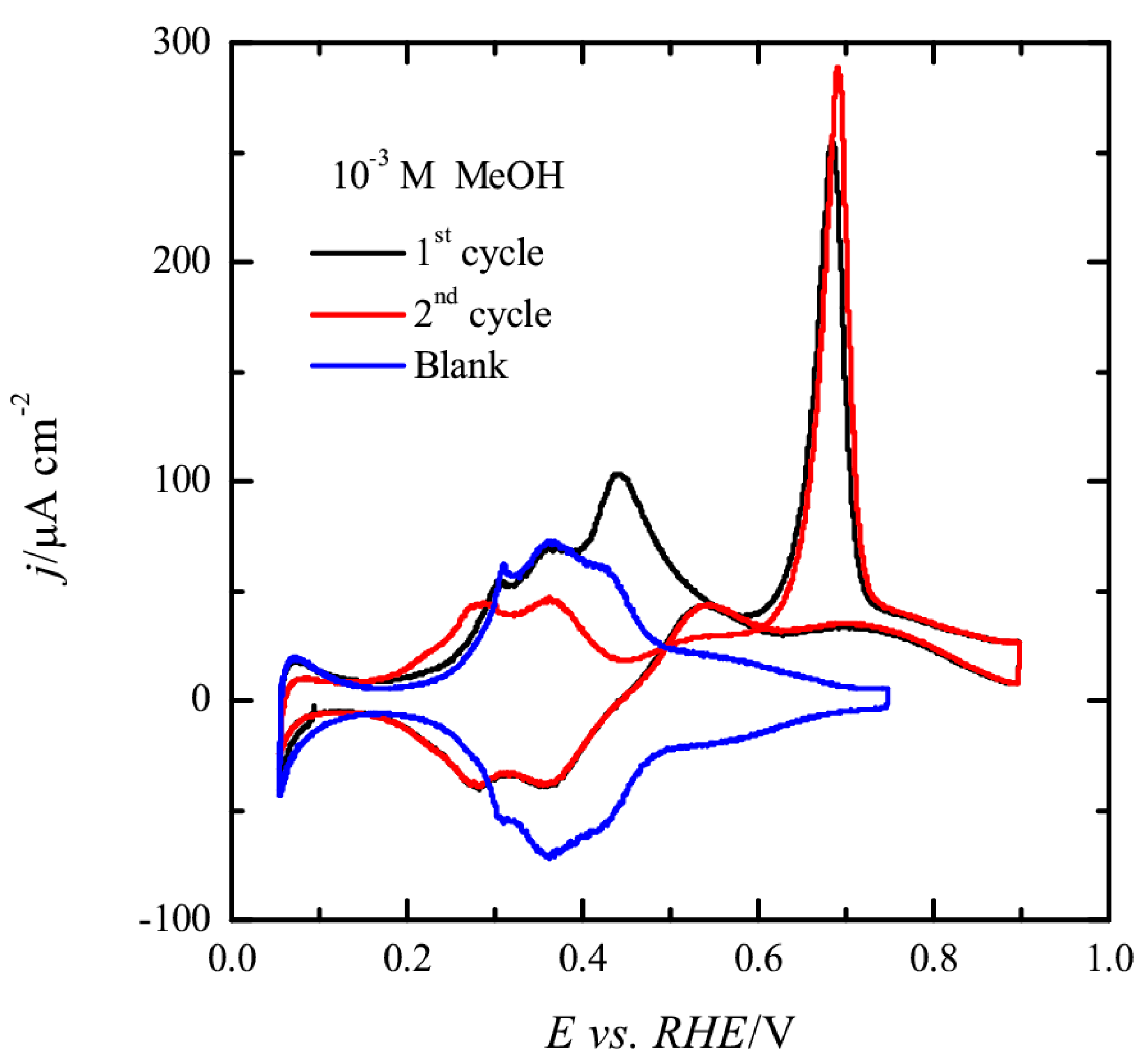
The process of CO formation on the electrode surface can be followed for the 10
−3 M methanol concentration (
Figure 7). As can be seen, the voltammogram for the first scan in the presence of 10
−3 M methanol overlaps with that recorded in the absence of methanol until 0.45 V, implying that no CO has been accumulated on the surface up to this potential value. Coinciding with the onset of OH adsorption, some additional currents are observed. These currents should correspond to the formation of CO from methanol according to the reaction
In fact, the analysis of the transient currents shows that CO is formed between 0.45 and 0.6 V [
14], implying that the formation of CO is only taking place in the region where OH is adsorbed. In the negative scan direction, some additional currents are observed in this region, which are also related to the formation of CO, as revealed by the significant diminution of the hydrogen adsorption charge observed below 0.4 V. After this first scan, the second scan does not show the currents at 0.45 V, because CO is already adsorbed on the surface. It should be noted that the following scans are identical to the second scan. For the 10
−2 M methanol concentration, the process of CO formation is not as visible as in 10
−3 M (inset of
Figure 6). While currents are recorded between 0.4 and 0.6 V, the peak is not well developed. It should be highlighted that, for this concentration, the results are very dependent on how the meniscus is formed. This fact implies that CO is being formed during the immersion of the electrode and the meniscus formation. While the electrode is immersed at a controlled potential (0.1 V), a few ms are required to establish the imposed potential. Due to the higher concentrations and the concomitant higher reaction rates, CO is being formed during this process. As can be seen in
Figure 6 for this concentration, hydrogen desorption states have been displaced to lower potential values and hydrogen desorption charge is smaller, which is the characteristic behavior of a Pt(100) surface partially covered by CO [
46]. Thus, a lower amount of CO can be formed in the positive scan, resulting in a less defined peak for this process.
Regarding the behavior of the methanol oxidation reaction on the Pt(100) electrode in different pH solutions (
Figure 8), the general behavior is the same as that recorded in 0.1 M HClO4. In the positive scan direction, the peak associated with the oxidation of the accumulated CO can be observed, followed by the presence of a second oxidation wave, which is also visible in the negative scan direction. In general, no significant variation of the currents is observed, and the measured currents in the broad wave in the negative scan direction are almost independent of the pH. The observed changes, especially in the shape of the peak associated with the oxidation of CO and the broad wave are probably related to the changes in the OH adsorption mode. The voltammograms in absence of methanol show a progressive evolution from the profile measured at pH = 1 (
Figure 7) to that obtained in alkaline solutions [
47]. While the OH coverage is not significantly affected by the pH, the differences in the voltammograms reflect that the energetics of the OH adsorption process is altered by pH [
47]. If, as proposed, OH involved in the reaction, the changes in the energetics should imply changes in the kinetics, that in this cases are translated in changes of the voltammetric shape. On the other hand, a reaction order cannot be obtained using this wave due to experimental problems. In this case, the raising part of the curve in the negative scan direction (that appearing between 0.6 and 0.7 V) should be used because it is not affected by the presence of adsorbed CO or any other inhibition process related to the formation of a compact OH layer. However, this region is very is very narrow and when curves are transformed in the SHE scale, the regions for the different pH do not overlap, preventing the accurate measurement of the current for the different pH values at a constant potential vs. SHE. Nevertheless, the observed behavior is similar to that observed for oxygen reduction, where currents are almost independent of the solution pH [
41].
For the Pt(110) electrode, the voltammogram for the oxidation of methanol shows a wave with a complex shape in the positive scan, whose onset is located at ca. 0.5 V in 10
−4 M methanol, whereas, in the negative scan direction, the currents are almost negligible (
Figure 9). These small currents cannot be assigned to the formation of adsorbed CO since the voltammetric profile in the low potential region overlaps with that measured in the absence of methanol. It should be noted that the signals appearing in the blank voltammogram between 0.06 and 0.30 V contain contributions both from hydrogen and OH adsorption. Then, the low currents recorded in the negative scan direction should be assigned to the oxidation of the surface, which is an irreversible process that occurs for this electrode above 0.8 V. As the concentration increases, the current also increases. However, the peak current difference between the positive and negative scan directions remains, due to the oxidation of the surface. It should be stressed that the oxidation of methanol is taking place, also on this electrode, on a surface partially covered by OH, reinforcing the connection between adsorbed OH and methanol oxidation. On the other hand, the solution pH effect on this surface is very similar to that described for the Pt(100) electrode. It should be noted that the voltammograms for this surface are very sensitive to the actual structure of the electrode, likely related to the fraction of the surface having the (1 × 2) reconstruction. This ratio depends, in turn, on the annealing conditions [
23], so that the measured currents for the methanol oxidation reaction for this electrode show large variability.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








