
Plasma Co-Polymerization of HMDSO and Limonene with an Atmospheric Pressure Plasma Jet

Abstract
:1. Introduction
2. Materials and Methods
2.1. Setup and Materials
2.2. Coating and Surface Analysis
3. Results and Discussion
3.1. Co-Polymerization of an Organic and an Organosilicon Precursor
3.2. Effects of Co-Polymerization on the Structure of the Siloxane Network
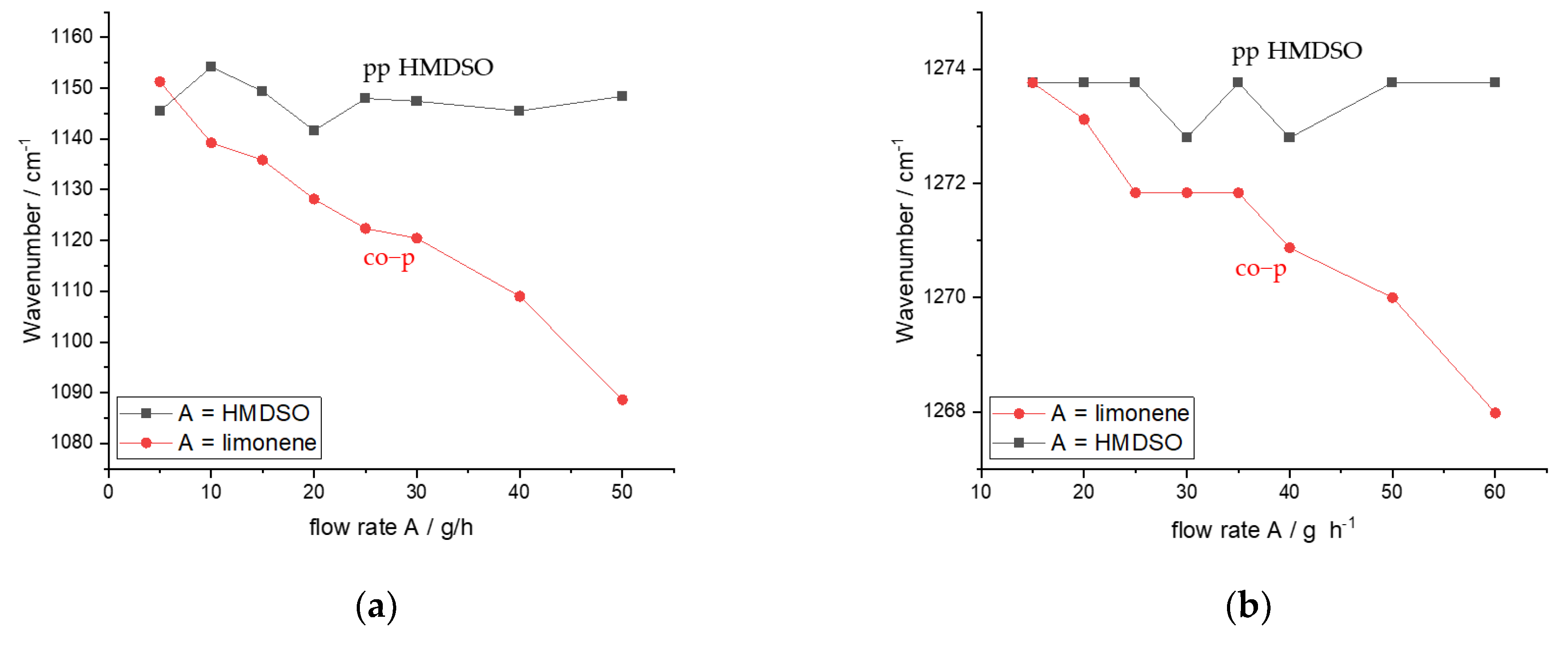
3.3. Individual Contribution of HMDSO and Lim to the Formation of the Co-p
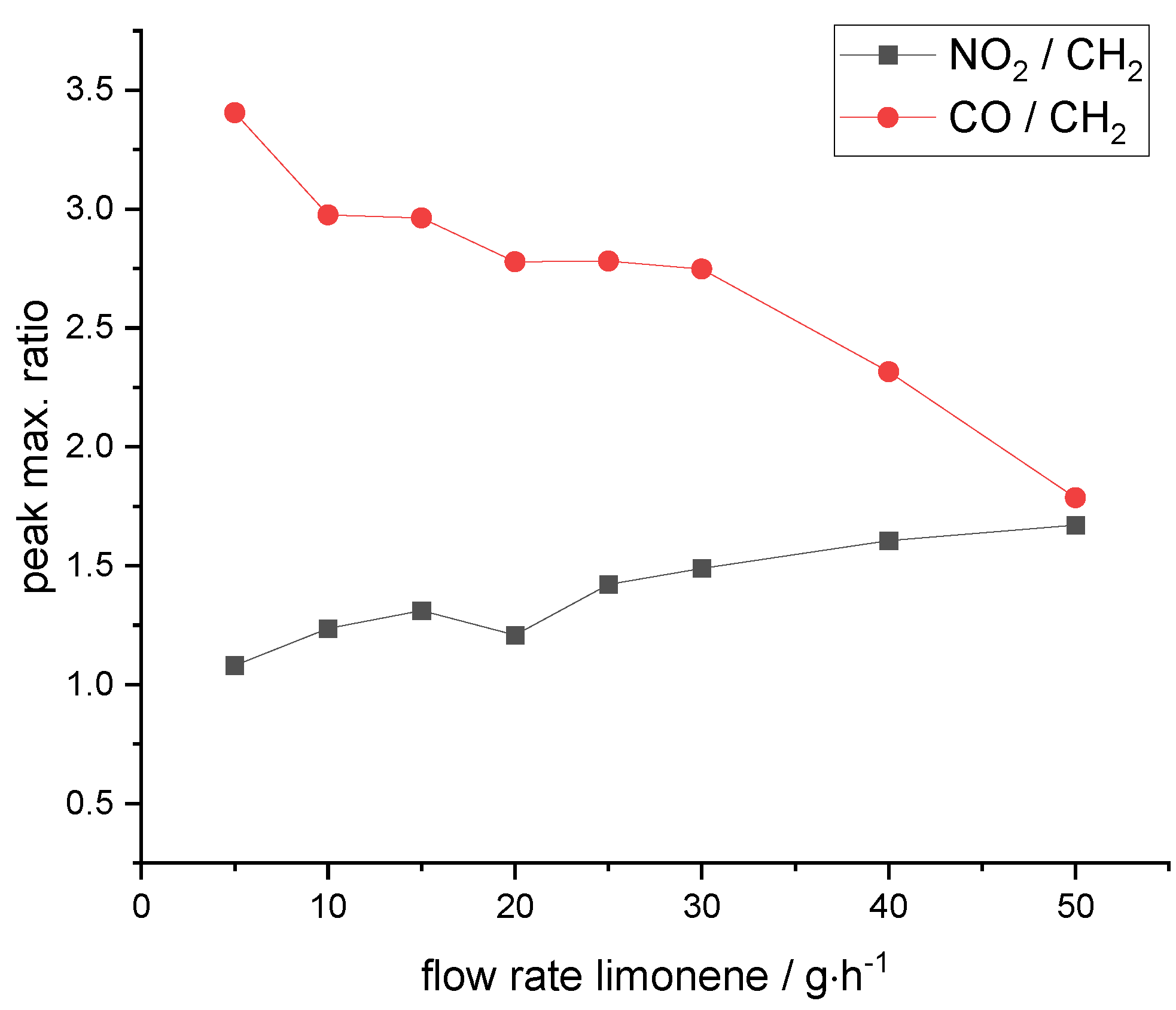
3.4. Role of Post-Plasma and In-Plasma Reactions in the Formation of Nitro and Carbonyl Functional Groups
4. Conclusions and Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fanelli, F.; Fracassi, F. Atmospheric pressure non-equilibrium plasma jet technology: General features, specificities and applications in surface processing of materials. Surf. Coat. Technol. 2017, 322, 174–201. [Google Scholar] [CrossRef]
- Merche, D.; Vandencasteele, N.; Reniers, F. Atmospheric plasmas for thin film deposition: A critical review. Thin Solid Films 2012, 520, 4219–4236. [Google Scholar] [CrossRef]
- Penkov, O.V.; Khadem, M.; Lim, W.-S.; Kim, D.-E. A review of recent applications of atmospheric pressure plasma jets for materials processing. J. Coat. Technol. Res. 2015, 12, 225–235. [Google Scholar] [CrossRef]
- Lommatzsch, U.; Ihde, J. Plasma Polymerization of HMDSO with an Atmospheric Pressure Plasma Jet for Corrosion Protection of Aluminum and Low-Adhesion Surfaces. Plasma Process. Polym. 2009, 6, 642–648. [Google Scholar] [CrossRef]
- Foest, R.; Kindel, E.; Ohl, A.; Stieber, M.; Weltmann, K.-D. Non-thermal atmospheric pressure discharges for surface modification. Plasma Phys. Control. Fusion 2005, 47, B525–B536. [Google Scholar] [CrossRef]
- Bringmann, P.; Rohr, O.; Gammel, F.J.; Jansen, I. Atmospheric Pressure Plasma Deposition of Adhesion Promotion Layers on Aluminium. Plasma Process. Polym. 2009, 6, S496–S502. [Google Scholar] [CrossRef]
- Pulpytel, J.; Kumar, V.; Peng, P.; Micheli, V.; Laidani, N.; Arefi-Khonsari, F. Deposition of Organosilicon Coatings by a Non-Equilibrium Atmospheric Pressure Plasma Jet: Design, Analysis and Macroscopic Scaling Law of the Process. Plasma Process. Polym. 2011, 8, 664–675. [Google Scholar] [CrossRef]
- Schäfer, J.; Foest, R.; Sigeneger, F.; Loffhagen, D.; Weltmann, K.-D.; Martens, U.; Hippler, R. Study of thin Film Formation From Silicon-Containing Precursors Produced by an RF Non-Thermal Plasma Jet at Atmospheric Pressure. Contrib. Plasma Phys. 2012, 52, 872–880. [Google Scholar] [CrossRef]
- Schäfer, J.; Foest, R.; Quade, A.; Ohl, A.; Weltmann, K.-D. Chemical Composition of SiO x Films Deposited by an Atmospheric Pressure Plasma Jet (APPJ). Plasma Process. Polym. 2009, 6, S519–S524. [Google Scholar] [CrossRef]
- Ramamoorthy, A.; Rahman, M.; Mooney, D.A.; Don MacElroy, J.M.; Dowling, D.P. The Influence of Process Parameters on Chemistry, Roughness and Morphology of Siloxane Films Deposited by an Atmospheric Plasma Jet System. Plasma Process. Polym. 2009, 6, S530–S536. [Google Scholar] [CrossRef]
- Castro, A.H.R.; Kodaira, F.V.; Prysiazhnyi, V.; Mota, R.P.; Kostov, K.G. Deposition of thin films using argon/acetylene atmospheric pressure plasma jet. Surf. Coat. Technol. 2017, 312, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Marcinauskas, L.; Grigonis, A.; Valatkevičius, P. Deposition of carbon structures at atmospheric pressure by plasma jet. J. Optoelectron. Adv. Mater. 2010, 12, 829. [Google Scholar]
- Van Vrekhem, S.; Morent, R.; de Geyter, N. Deposition of a PMMA coating with an atmospheric pressure plasma jet. J. Coat. Technol. Res. 2018, 15, 679–690. [Google Scholar] [CrossRef]
- Yan, J.; Hosoi, K.; Kuroda, S. Polymerization of Maleic Anhydride Induced by Non-Equilibrium Atmospheric Pressure Argon Plasma. AMM 2014, 670–671, 244–248. [Google Scholar] [CrossRef]
- Yasuda, H. Glow discharge polymerization. In Thin Film Processes; Academic Press: San Diego, CA, USA, 1978. [Google Scholar]
- Kakaroglou, A.; Scheltjens, G.; Nisol, B.; de Graeve, I.; van Assche, G.; van Mele, B.; Willem, R.; Biesemanns, M.; Reniers, F.; Terryn, H. Deposition and Characterisation of Plasma Polymerised Allyl Methacrylate Based Coatings. Plasma Process. Polym. 2012, 9, 799–807. [Google Scholar] [CrossRef]
- Petersen, J.; Becker, C.; Fouquet, T.; Addiego, F.; Toniazzo, V.; Dinia, A.; Ruch, D. Nano-ordered thin films achieved by soft atmospheric plasma polymerization. RSC Adv. 2013, 3, 4416. [Google Scholar] [CrossRef]
- Petersen, J.; Bechara, R.; Bardon, J.; Fouquet, T.; Ziarelli, F.; Daheron, L.; Ball, V.; Toniazzo, V.; Michel, M.; Dinia, A.; et al. Atmospheric Plasma Deposition Process: A Versatile Tool for the Design of Tunable Siloxanes-Based Plasma Polymer Films. Plasma Process. Polym. 2011, 8, 895–903. [Google Scholar] [CrossRef]
- Friedrich, J. Mechanisms of Plasma Polymerization—Reviewed from a Chemical Point of View. Plasma Process. Polym. 2011, 8, 783–802. [Google Scholar] [CrossRef]
- Lommatzsch, U.; Pasedag, D.; Baalmann, A.; Ellinghorst, G.; Wagner, H.-E. Atmospheric Pressure Plasma Jet Treatment of Polyethylene Surfaces for Adhesion Improvement. Plasma Process. Polym. 2007, 4, S1041–S1045. [Google Scholar] [CrossRef]
- Thomas, M.; von Hausen, M.; Klages, C.-P.; Baumhof, P. Generation of stable coatings with carboxylic groups by copolymerization of MAA and VTMS using DBD at atmospheric pressure. Plasma Process. Polym. 2007, 4, 475–481. [Google Scholar] [CrossRef]
- Marcinauskas, L.; Valinčius, V.; Grigonis, A. Deposition and structure characterization of carbon films prepared at atmospheric pressure by plasma jet. Surf. Coat. Technol. 2011, 205, 71–74. [Google Scholar] [CrossRef]
- Han, G.-J.; Chung, S.-N.; Chun, B.-H.; Kim, C.-K.; Oh, K.H.; Cho, B.-H. 1,3-Butadiene as an Adhesion Promoter Between Composite Resin and Dental Ceramic in a Dielectric Barrier Discharge Jet. Plasma Chem. Plasma Process. 2013, 33, 539–551. [Google Scholar] [CrossRef]
- Ben Said, S.; Arefi-Khonsari, F.; Pulpytel, J. Plasma Polymerization of 3-Aminopropyltrietoxysilane (APTES) by Open-Air Atmospheric Arc Plasma Jet for In-Line Treatments. Plasma Process. Polym. 2016, 13, 1025–1035. [Google Scholar] [CrossRef]
- Alba-Elías, F.; Sainz-García, E.; González-Marcos, A.; Ordieres-Meré, J. Tribological behavior of plasma-polymerized aminopropyltriethoxysilane films deposited on thermoplastic elastomers substrates. Thin Solid Films 2013, 540, 125–134. [Google Scholar] [CrossRef]
- Grill, A. Porous pSiCOH Ultralow- k Dielectrics for Chip Interconnects Prepared by PECVD. Annu. Rev. Mater. Res. 2009, 39, 49–69. [Google Scholar] [CrossRef]
- Ding, Z.-J.; Ding, S.-J.; Zhang, W. Plasma enhanced chemical vapor deposition ultralow dielectric constant films using triethoxymethylsilane and limonene as precursors. In Proceedings of the 2014 12th IEEE International Conference on Solid-State and Integrated Circuit Technology (ICSICT), Guilin, China, 28–31 October 2014; pp. 1–3. [Google Scholar]
- Cho, S.-J.; Bae, I.-S.; Boo, J.-H.; Lee, S.; Jung, D. The Optical and Mechanical Properties of Toluene-TEOS Hybrid Plasma-Polymer Thin Films Deposited by PECVD. J. Korean Phy. Soc. 2009, 55, 1780–1784. [Google Scholar] [CrossRef]
- Beck, A.J.; Jones, F.R.; Short, R.D. Plasma copolymerization as a route to the fabrication of new surfaces with controlled amounts of specific chemical functionality. Polymer 1996, 37, 5537–5539. [Google Scholar] [CrossRef]
- Grill, A.; Neumayer, D.A. Structure of low dielectric constant to extreme low dielectric constant SiCOH films: Fourier transform infrared spectroscopy characterization. J. Appl. Phys. 2003, 94, 6697–6707. [Google Scholar] [CrossRef]
- Manakhov, A.; Moreno-Couranjou, M.; Choquet, P.; Boscher, D.; Pireaux, J.-J. Diene functionalisation of atmospheric plasma copolymer thin films. Surf. Coat. Technol. 2011, 205, 466–469. [Google Scholar] [CrossRef]
- Abessolo Ondo, D.; Loyer, F.; Chemin, J.-B.; Bulou, S.; Choquet, P.; Boscher, N.D. Atmospheric plasma oxidative polymerization of ethylene dioxythiophene (EDOT) for the large-scale preparation of highly transparent conducting thin films. Plasma Process. Polym. 2018, 15, 1700172. [Google Scholar] [CrossRef] [Green Version]
- Makhneva, E.; Farka, Z.; Pastucha, M.; Obrusník, A.; Horáčková, V.; Skládal, P.; Zajíčková, L. Maleic anhydride and acetylene plasma copolymer surfaces for SPR immunosensing. Anal. Bioanal. Chem. 2019, 411, 7689–7697. [Google Scholar] [CrossRef] [PubMed]
- Manakhov, A.; Michlíček, M.; Nečas, D.; Polčák, J.; Makhneva, E.; Eliáš, M.; Zajíčková, L. Carboxyl-rich coatings deposited by atmospheric plasma co-polymerization of maleic anhydride and acetylene. Surf. Coat. Technol. 2016, 295, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Manakhov, A.; Moreno-Couranjou, M.; Choquet, P.; Boscher, D.; Pireaux, J.-J. Atmospheric Pressure Pulsed Plasma Copolymerisation of Maleic Anhydride and Vinyltrimethoxysilane: Influence of Electrical Parameters on Chemistry, Morphology and Deposition Rate of the Coatings. Plasma Process. Polym. 2012, 9, 435–445. [Google Scholar] [CrossRef]
- Jang, H.J.; Park, C.-S.; Jung, E.Y.; Bae, G.T.; Shin, B.J.; Tae, H.-S. Synthesis and Properties of Thiophene and Aniline Copolymer Using Atmospheric Pressure Plasma Jets Copolymerization Technique. Polymers 2020, 12, 2225. [Google Scholar] [CrossRef]
- Ben Salem, D.; Carton, O.; Fakhouri, H.; Pulpytel, J.; Arefi-Khonsari, F. Deposition of Water Stable Plasma Polymerized Acrylic Acid/MBA Organic Coatings by Atmospheric Pressure Air Plasma Jet. Plasma Process. Polym. 2014, 11, 269–278. [Google Scholar] [CrossRef]
- Hossain, M.M.; Trinh, Q.H.; Nguyen, D.B.; Sudhakaran, M.S.P.; Mok, Y.S. Robust hydrophobic coating on glass surface by an atmospheric-pressure plasma jet for plasma-polymerisation of hexamethyldisiloxane conjugated with (3-aminopropyl) triethoxysilane. Surf. Eng. 2019, 35, 466–475. [Google Scholar] [CrossRef]
- Gilliam, M.A.; Farhat, S.A.; Garner, G.E.; Stubbs, B.P.; Peterson, B.B. Characterization of the deposition behavior and changes in bonding structures of hexamethyldisiloxane and decamethylcyclopentasiloxane atmospheric plasma-deposited films*. Plasma Process. Polym. 2019, 16, e1900024. [Google Scholar] [CrossRef]
- Grill, A. Plasma enhanced chemical vapor deposited SiCOH dielectrics: From low-k to extreme low-k interconnect materials. J. Appl. Phys. 2003, 93, 1785–1790. [Google Scholar] [CrossRef]
- Spektroskopische Methoden in der Organischen Chemie, 7th ed.; Hesse, M.; Meier, H.; Zeeh, B. (Eds.) Thieme: Stuttgart, Germany, 2005. [Google Scholar]
- Prysiazhnyi, V.; Kratochvil, J.; Stranak, V. Tailored wettability of plasma polymers made of C–F, C–H, and N–H. Plasma Process. Polym. 2019, 16, 1900076. [Google Scholar] [CrossRef]
- Kim, C.Y.; Kim, S.H.; Navamathavan, R.; Choi, C.K.; Jeung, W.Y. Characteristics of low-k SiOC(–H) films deposited at various substrate temperature by PECVD using DMDMS/O2 precursor. Thin Solid Films 2007, 516, 340–344. [Google Scholar] [CrossRef]
- Hegemann, D.; Nisol, B.; Gaiser, S.; Watson, S.; Wertheimer, M.R. Energy conversion efficiency in low- and atmospheric-pressure plasma polymerization processes with hydrocarbons. Phys. Chem. Chem. Phys. 2019, 21, 8698–8708. [Google Scholar] [CrossRef] [PubMed]
- Enache, I.; Caquineau, H.; Gherardi, N.; Paulmier, T.; Maechler, L.; Massines, F. Transport Phenomena in an Atmospheric-Pressure Townsend Discharge Fed by N2/N2O/HMDSO Mixtures. Plasma Process. Polym. 2007, 4, 806–814. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Babayan, S.E.; Tu, V.J.; Park, J.; Henins, I.; Hicks, R.F.; Selwyn, G.S. Etching materials with an atmospheric-pressure plasma jet. Plasma Sources Sci. Technol. 1998, 7, 282–285. [Google Scholar] [CrossRef]
- Wissenschaftliche Veröffentlichungen aus dem Siemens-Konzern; Erster Band 1920–1922; Becker, H.; Boedeker, K.; von Buol, H.; Ebeling, A.; Engelhardt, V.; Erlwein, G.; Fellinger, R.; Feuerlein, O.; Franke, A.; Friese, M.; et al. (Eds.) Springer: Berlin/Heidelberg, Germany, 1922. [Google Scholar]
- Hashmi, S. (Ed.) Comprehensive Materials Processing; Elsevier: Amsterdam, The Netherlands, 2014; Volume 13. [Google Scholar]
- Yumitori, S. Correlation of C1s chemical state intensities with the O1s intensity in the XPS analysis of anodically oxidized glass-like carbon samples. J. Mater. Sci. 2000, 35, 139–146. [Google Scholar] [CrossRef]
- Truica-Marasescu, F.; Wertheimer, M.R. Nitrogen-Rich Plasma-Polymer Films for Biomedical Applications. Plasma Process. Polym. 2008, 5, 44–57. [Google Scholar] [CrossRef]
- Finke, B.; Schröder, K.; Ohl, A. Surface Radical Detection on NH3-Plasma Treated Polymer Surfaces Using the Radical Scavenger NO. Plasma Process. Polym. 2008, 5, 386–396. [Google Scholar] [CrossRef]
- Schmidt-Bleker, A.; Bansemer, R.; Reuter, S. How to produce an NOx- instead of Ox-based chemistry with a cold atmospheric plasma jet. Plasma Process. Polym. 2016, 13, 1120–1127. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coating | XPS Chemical Composition | |||
|---|---|---|---|---|
| at.-% | ||||
| Carbon | Oxygen | Silicon | Nitrogen | |
| pp HMDSO | 25.4 | 46.1 | 28.3 | 0.3 |
| co-p | 42.7 | 39.4 | 17.4 | 0.7 |
| pp lim | 71.2 | 27.1 | 0.2 | 1.6 |
| pp lim N2 | 70.2 | 23.2 | 0.1 | 6.6 |
| theoretical co-p | 48.3 | 36.6 | 14.3 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wulf, G.; Mayer, B.; Lommatzsch, U. Plasma Co-Polymerization of HMDSO and Limonene with an Atmospheric Pressure Plasma Jet. Plasma 2022, 5, 44-59. https://doi.org/10.3390/plasma5010004
Wulf G, Mayer B, Lommatzsch U. Plasma Co-Polymerization of HMDSO and Limonene with an Atmospheric Pressure Plasma Jet. Plasma. 2022; 5(1):44-59. https://doi.org/10.3390/plasma5010004
Chicago/Turabian StyleWulf, Gerrit, Bernd Mayer, and Uwe Lommatzsch. 2022. "Plasma Co-Polymerization of HMDSO and Limonene with an Atmospheric Pressure Plasma Jet" Plasma 5, no. 1: 44-59. https://doi.org/10.3390/plasma5010004