
Effect of Water and Salt on the Colloidal Stability of Latex Particles in Ionic Liquid Solutions


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Dynamic Light Scattering
2.3. Phase Analysis Light Scattering
3. Results and Discussion
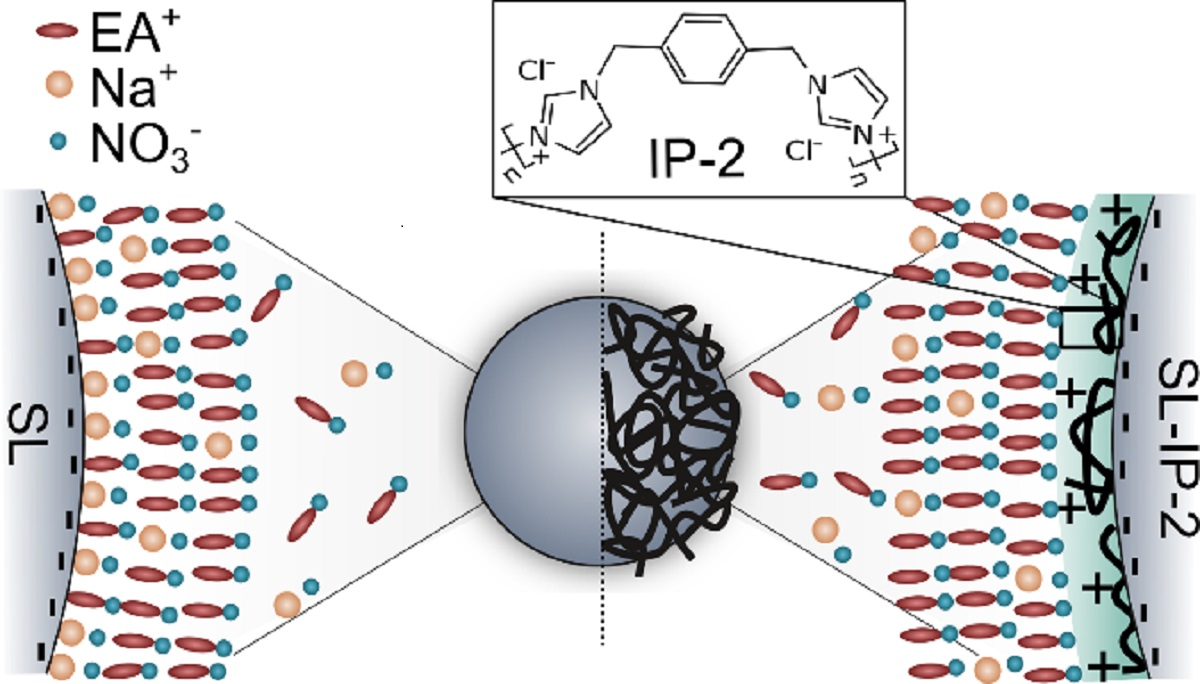
3.1. IP-2 Functionalization of SL
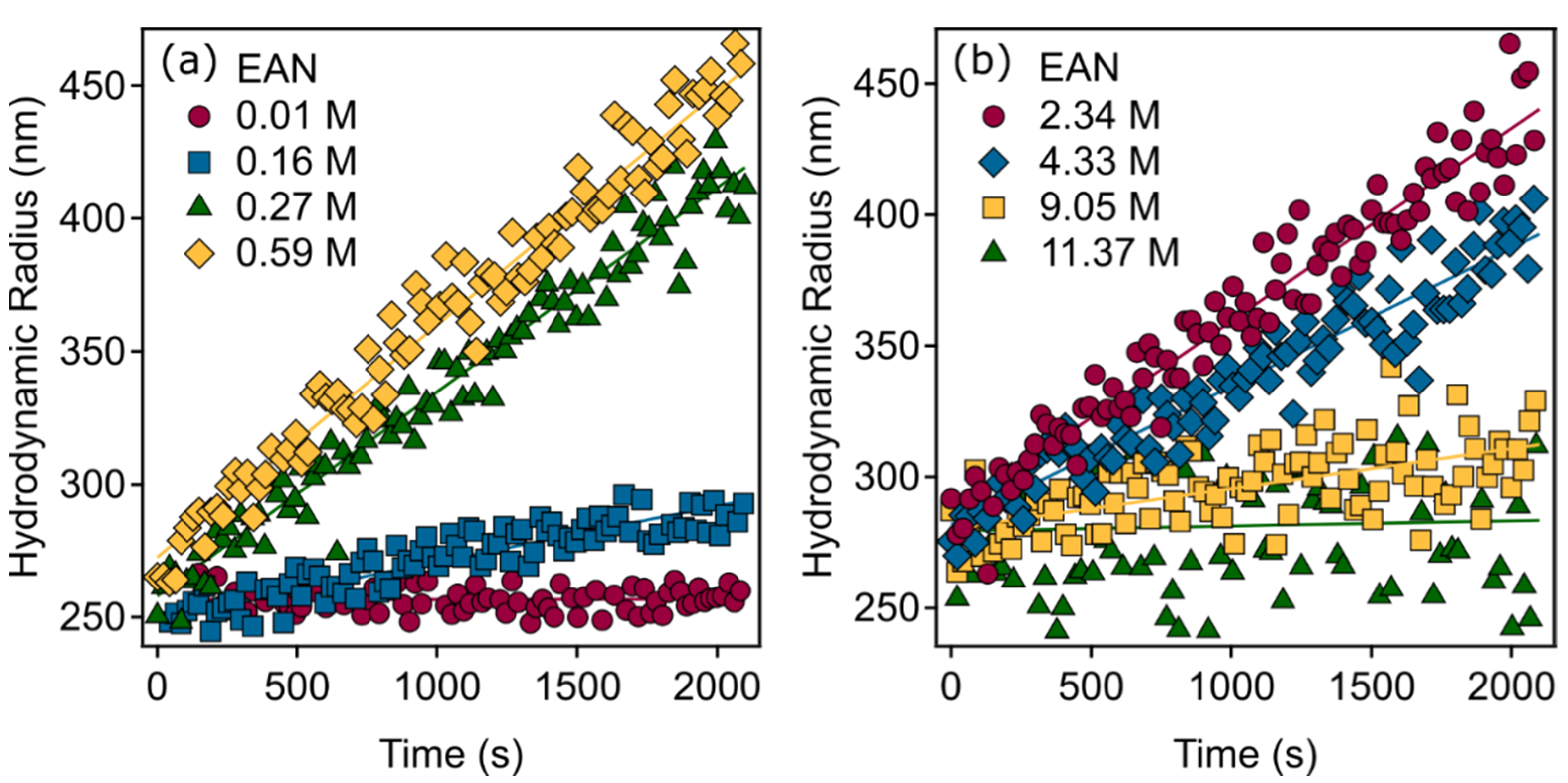
3.2. Generic Trend in Aggregation Kinetics
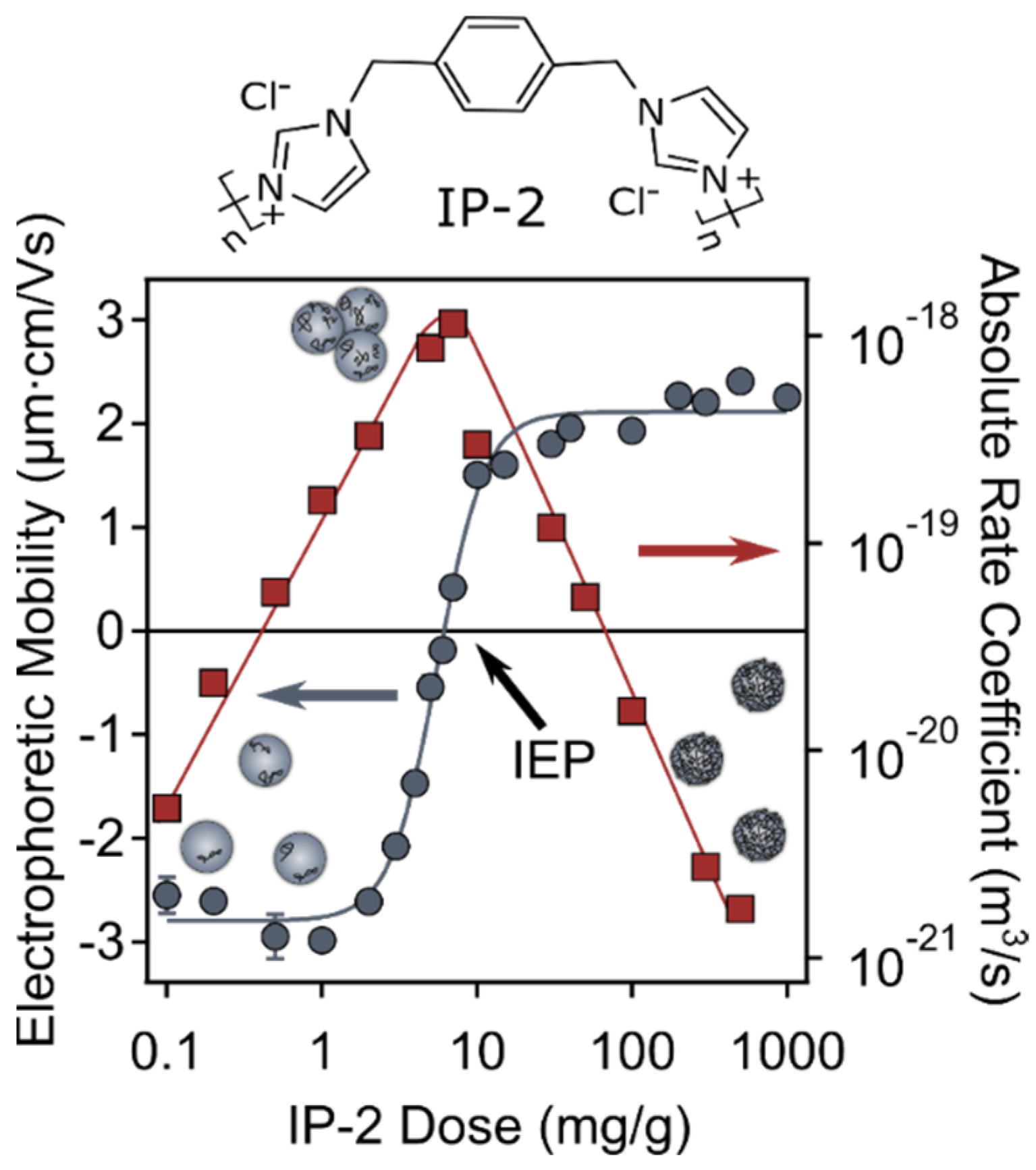
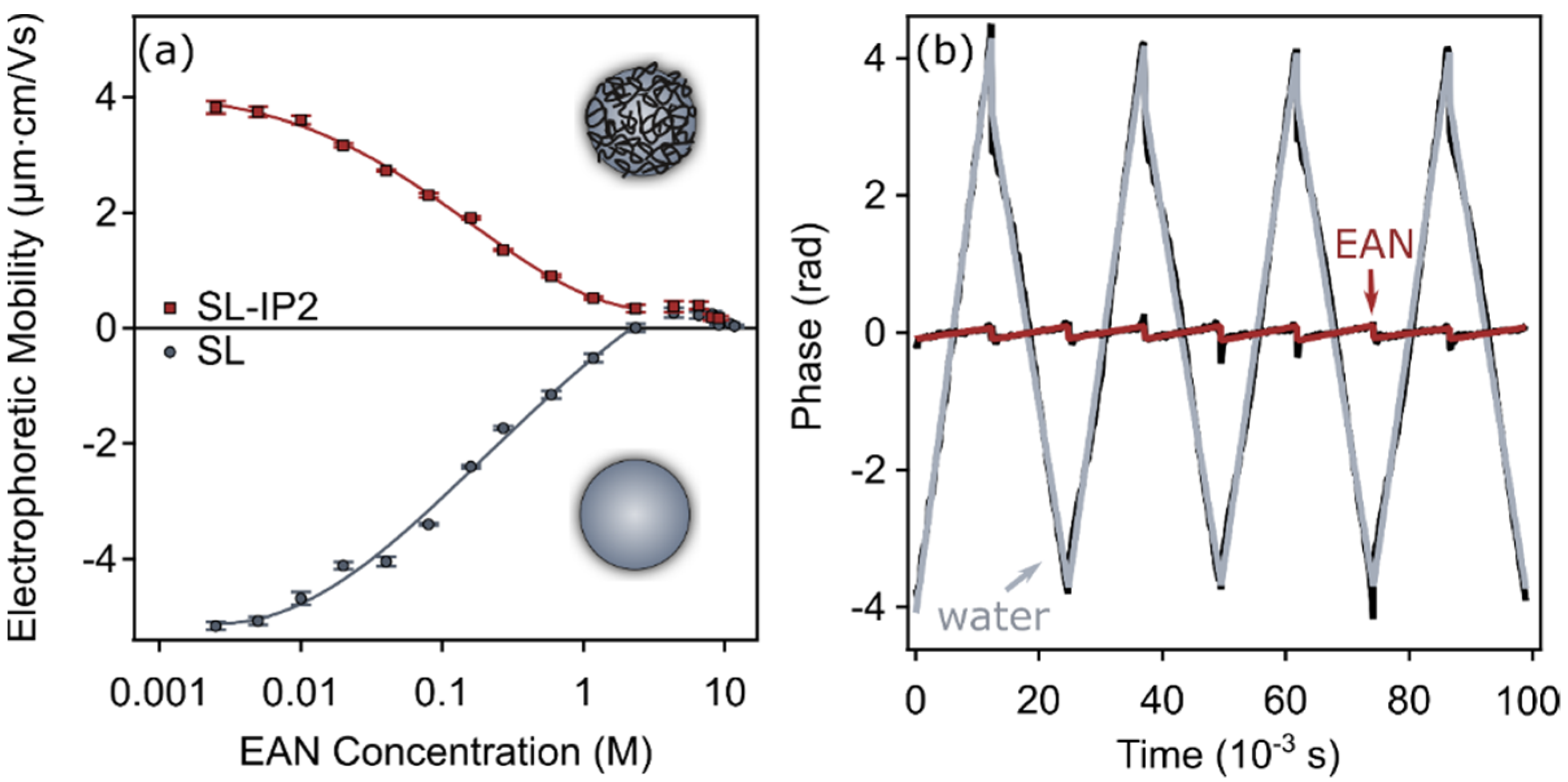
3.3. Charging Characteristics
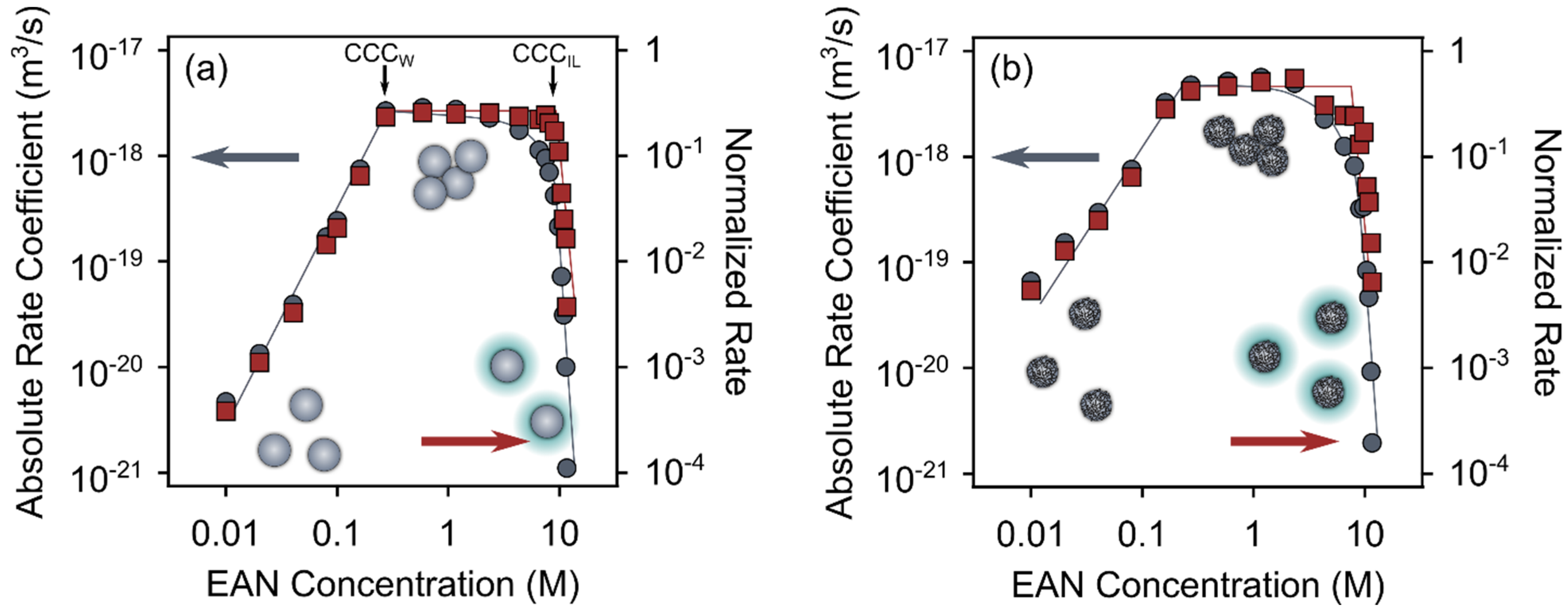
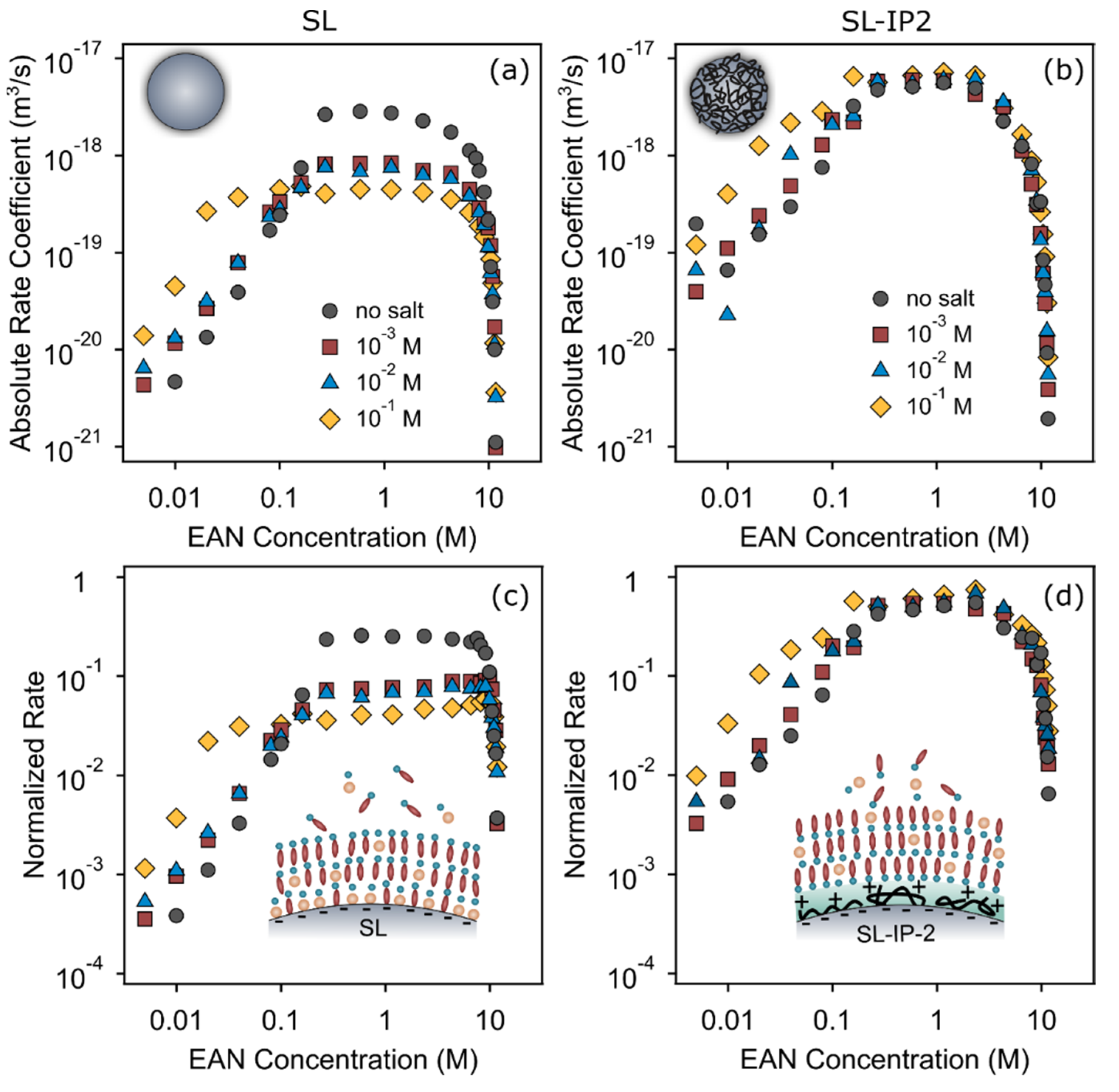
3.4. Effect of Added Salt
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Armand, M.; Endres, F.; MacFarlane, D.R.; Ohno, H.; Scrosati, B. Ionic-liquid materials for the electrochemical challenges of the future. Nat. Mater. 2009, 8, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Donato, K.Z.; Matejka, L.; Mauler, R.S.; Donato, R.K. Recent applications of ionic liquids in the sol-gel process for polymer–silica nanocomposites with ionic interfaces. Colloids Interfaces 2017, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Franca, J.M.P.; Vieira, S.I.C.; Lourenco, M.J.V.; Murshed, S.M.S.; de Castro, C.A.N. Thermal conductivity of C4mim (CF3SO2)2N and C2mim EtSO4 and their ionanofluids with carbon nanotubes: Experiment and theory. J. Chem. Eng. Data 2013, 58, 467–476. [Google Scholar] [CrossRef]
- Minea, M.A.; Murshed, S.M.S. Ionic liquids-based nanocolloids—A review of progress and prospects in convective heat transfer applications. Nanomaterials 2021, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- He, Z.Q.; Alexandridis, P. Ionic liquid and nanoparticle hybrid systems: Emerging applications. Adv. Colloid Interface Sci. 2017, 244, 54–70. [Google Scholar] [CrossRef]
- Ueno, K.; Watanabe, M. From colloidal stability in ionic liquids to advanced soft materials using unique media. Langmuir 2011, 27, 9105–9115. [Google Scholar] [CrossRef]
- Tunckol, M.; Durand, J.; Serp, P. Carbon nanomaterial-ionic liquid hybrids. Carbon 2012, 50, 4303–4334. [Google Scholar] [CrossRef]
- Parvulescu, V.I.; Hardacre, C. Catalysis in ionic liquids. Chem. Rev. 2007, 107, 2615–2665. [Google Scholar] [CrossRef]
- Hallett, J.P.; Welton, T. Room-temperature ionic liquids: Solvents for synthesis and catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef]
- Taskin, M.; Cognigni, A.; Zirbs, R.; Reimhult, E.; Bica, K. Surface-active ionic liquids for palladium-catalysed cross coupling in water: Effect of ionic liquid concentration on the catalytically active species. RSC Adv. 2017, 7, 41144–41151. [Google Scholar] [CrossRef] [Green Version]
- Le Bideau, J.; Viau, L.; Vioux, A. Ionogels, ionic liquid based hybrid materials. Chem. Soc. Rev. 2011, 40, 907–925. [Google Scholar] [CrossRef] [PubMed]
- Silvester, D.S.; Jamil, R.; Doblinger, S.; Zhang, Y.X.; Atkin, R.; Li, H. Electrical double layer structure in ionic liquids and its importance for supercapacitor, battery, sensing, and lubrication applications. J. Phys. Chem. C 2021, 125, 13707–13720. [Google Scholar] [CrossRef]
- Beattie, D.A.; Espinosa-Marzal, R.M.; Ho, T.T.M.; Popescu, M.N.; Ralston, J.; Richard, C.J.E.; Sellapperumage, P.M.F.; Krasowska, M. Molecularly-thin precursor films of imidazolium-based ionic liquids on mica. J. Phys. Chem. C 2013, 117, 23676–23684. [Google Scholar] [CrossRef]
- Mezger, M.; Schroder, H.; Reichert, H.; Schramm, S.; Okasinski, J.S.; Schoder, S.; Honkimaki, V.; Deutsch, M.; Ocko, B.M.; Ralston, J.; et al. Molecular layering of fluorinated ionic liquids at a charged sapphire (0001) surface. Science 2008, 322, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Elimelech, M.; Gregory, J.; Jia, X.; Williams, R.A. Particle Deposition and Aggregation: Measurement, Modeling, and Simulation; Butterworth-Heinemann Ltd.: Oxford, UK, 1995. [Google Scholar]
- Israelachvili, J. Intermolecular and Surface Forces, 3rd ed.; Academic Press: London, UK, 2011. [Google Scholar]
- Russel, W.B.; Saville, D.A.; Schowalter, W.R. Colloidal Dispersions; Cambridge University Press: Cambridge, UK, 1989. [Google Scholar]
- Ueno, K.; Inaba, A.; Kondoh, M.; Watanabe, M. Colloidal stability of bare and polymer-grafted silica nanoparticles in ionic liquids. Langmuir 2008, 24, 5253–5259. [Google Scholar] [CrossRef] [PubMed]
- Asencio, R.A.; Cranston, E.D.; Atkin, R.; Rutland, M.W. Ionic liquid nanotribology: Stiction suppression and surface induced shear thinning. Langmuir 2012, 28, 9967–9976. [Google Scholar] [CrossRef]
- Hjalmarsson, N.; Atkin, R.; Rutland, M.W. Is the boundary layer of an ionic liquid equally lubricating at higher temperature? Phys. Chem. Chem. Phys. 2016, 18, 9232–9239. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Werzer, O.; Webber, G.B.; Warr, G.G.; Atkin, R. Surprising particle stability and rapid sedimentation rates in an ionic liquid. J. Phys. Chem. Lett. 2010, 1, 64–68. [Google Scholar] [CrossRef]
- Szilagyi, I.; Szabo, T.; Desert, A.; Trefalt, G.; Oncsik, T.; Borkovec, M. Particle aggregation mechanisms in ionic liquids. Phys. Chem. Chem. Phys. 2014, 16, 9515–9524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and nanostructure in ionic liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.; Imberti, S.; Warr, G.G.; Atkin, R. The nature of hydrogen bonding in protic ionic liquids. Angew. Chem. Int. Ed. 2013, 52, 4623–4627. [Google Scholar] [CrossRef]
- Hao, J.C.; Zemb, T. Self-assembled structures and chemical reactions in room-temperature ionic liquids. Curr. Opin. Colloid Interface Sci. 2007, 12, 129–137. [Google Scholar] [CrossRef]
- Werzer, O.; Cranston, E.D.; Warr, G.G.; Atkin, R.; Rutland, M.W. Ionic liquid nanotribology: Mica-silica interactions in ethylammonium nitrate. Phys. Chem. Chem. Phys. 2012, 14, 5147–5152. [Google Scholar] [CrossRef] [PubMed]
- Elbourne, A.; Voitchovsky, K.; Warr, G.G.; Atkin, R. Ion structure controls ionic liquid near-surface and interfacial nanostructure. Chem. Sci. 2015, 6, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Sheehan, A.; Jurado, L.A.; Ramakrishna, S.N.; Arcifa, A.; Rossi, A.; Spencer, N.D.; Espinosa-Marzal, R.M. Layering of ionic liquids on rough surfaces. Nanoscale 2016, 8, 4094–4106. [Google Scholar] [CrossRef] [PubMed]
- Oncsik, T.; Desert, A.; Trefalt, G.; Borkovec, M.; Szilagyi, I. Charging and aggregation of latex particles in aqueous solutions of ionic liquids: Towards an extended Hofmeister series. Phys. Chem. Chem. Phys. 2016, 18, 7511–7520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katana, B.; Takács, D.; Csapo, E.; Szabo, T.; Jamnik, A.; Szilagyi, I. Ion specific effects on the stability of halloysite nanotube colloids—Inorganic salts versus ionic liquids. J. Phys. Chem. B 2020, 124, 9757–9765. [Google Scholar] [CrossRef]
- Katana, B.; Takács, D.; Szerlauth, A.; Sáringer, S.; Varga, G.; Jamnik, A.; Bobbink, F.D.; Dyson, P.J.; Szilagyi, I. Aggregation of halloysite nanotubes in the presence of multivalent ions and ionic liquids. Langmuir 2021, 37, 11869–11879. [Google Scholar] [CrossRef] [PubMed]
- Riedl, J.C.; Kazemi, M.A.A.; Cousin, F.; Dubois, E.; Fantini, S.; Lois, S.; Perzynski, R.; Peyre, V. Colloidal dispersions of oxide nanoparticles in ionic liquids: Elucidating the key parameters. Nanoscale Adv. 2020, 2, 1560–1572. [Google Scholar] [CrossRef] [Green Version]
- Nordstrom, J.; Aguilera, L.; Matic, A. Effect of lithium salt on the stability of dispersions of fumed silica in the ionic liquid BMImBF4. Langmuir 2012, 28, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Han, M.W.; Espinosa-Marzal, R.M. Influence of water on structure, dynamics, and electrostatics of hydrophilic and hydrophobic ionic liquids in charged and hydrophilic confinement between mica surfaces. ACS Appl. Mater. Interfaces 2019, 11, 33465–33477. [Google Scholar] [CrossRef]
- Hayes, R.; Borisenko, N.; Corr, B.; Webber, G.B.; Endres, F.; Atkin, R. Effect of dissolved LiCl on the ionic liquid-Au(111) electrical double layer structure. Chem. Commun. 2012, 48, 10246–10248. [Google Scholar] [CrossRef]
- Katana, B.; Takács, D.; Bobbink, F.D.; Dyson, P.J.; Alsharif, N.B.; Tomšič, M.; Szilagyi, I. Masking specific effects of ionic liquid constituents at the solid–liquid interface by surface functionalization. Phys. Chem. Chem. Phys. 2020, 22, 24764–24770. [Google Scholar] [CrossRef]
- Takács, D.; Katana, B.; Szerlauth, A.; Sebők, D.; Tomšič, M.; Szilagyi, I. Influence of adsorption of ionic liquid constituents on the stability of layered double hydroxide colloids. Soft Matter 2021, 17, 9116–9124. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Bobbink, F.D.; Fei, Z.F.; Dyson, P.J. Polyimidazolium salts: Robust catalysts for the cycloaddition of carbon dioxide into carbonates in solvent-free conditions. ChemSusChem 2017, 10, 2728–2735. [Google Scholar] [CrossRef] [PubMed]
- Zarrougui, R.; Dhahbi, M.; Lemordant, D. Transport and thermodynamic properties of ethylammonium nitrate-water binary mixtures: Effect of temperature and composition. J. Solut. Chem. 2015, 44, 686–702. [Google Scholar] [CrossRef]
- Holthoff, H.; Egelhaaf, S.U.; Borkovec, M.; Schurtenberger, P.; Sticher, H. Coagulation rate measurements of colloidal particles by simultaneous static and dynamic light scattering. Langmuir 1996, 12, 5541–5549. [Google Scholar] [CrossRef]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering; Robert E. Krieger Publishing: Malabar, FL, USA, 1990. [Google Scholar]
- Cao, T.C.; Borkovec, M.; Trefalt, G. Heteroaggregation and homoaggregation of latex particles in the presence of alkyl sulfate surfactants. Colloids Interfaces 2020, 4, 52. [Google Scholar] [CrossRef]
- Yu, W.L.; Matijevic, E.; Borkovec, M. Absolute heteroaggregation rate constants by multiangle static and dynamic light scattering. Langmuir 2002, 18, 7853–7860. [Google Scholar] [CrossRef]
- Hierrezuelo, J.; Vaccaro, A.; Borkovec, M. Stability of negatively charged latex particles in the presence of a strong cationic polyelectrolyte at elevated ionic strengths. J. Colloid Interface Sci. 2010, 347, 202–208. [Google Scholar] [CrossRef]
- Rouster, P.; Dondelinger, M.; Galleni, M.; Nysten, B.; Jonas, A.M.; Glinel, K. Layer-by-layer assembly of enzyme-loaded halloysite nanotubes for the fabrication of highly active coatings. Colloid Surf. B 2019, 178, 508–514. [Google Scholar] [CrossRef]
- Trefalt, G.; Szilagyi, I.; Borkovec, M. Poisson-Boltzmann description of interaction forces and aggregation rates involving charged colloidal particles in asymmetric electrolytes. J. Colloid Interface Sci. 2013, 406, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Oncsik, T.; Trefalt, G.; Borkovec, M.; Szilagyi, I. Specific ion effects on particle aggregation induced by monovalent salts within the Hofmeister series. Langmuir 2015, 31, 3799–3807. [Google Scholar] [CrossRef] [PubMed]
- Oncsik, T.; Trefalt, G.; Csendes, Z.; Szilagyi, I.; Borkovec, M. Aggregation of negatively charged colloidal particles in the presence of multivalent cations. Langmuir 2014, 30, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Valmacco, V.; Trefalt, G.; Maroni, P.; Borkovec, M. Direct force measurements between silica particles in aqueous solutions of ionic liquids containing 1-butyl-3-methylimidazolium (BMIM). Phys. Chem. Chem. Phys. 2015, 17, 16553–16559. [Google Scholar] [CrossRef]
- Sinha, P.; Szilagyi, I.; Ruiz-Cabello, F.J.M.; Maroni, P.; Borkovec, M. Attractive forces between charged colloidal particles induced by multivalent ions revealed by confronting aggregation and direct force measurements. J. Phys. Chem. Lett. 2013, 4, 648–652. [Google Scholar] [CrossRef]
- Kobayashi, M.; Juillerat, F.; Galletto, P.; Bowen, P.; Borkovec, M. Aggregation and charging of colloidal silica particles: Effect of particle size. Langmuir 2005, 21, 5761–5769. [Google Scholar] [CrossRef]
- Vanecht, E.; Binnemans, K.; Patskovsky, S.; Meunier, M.; Seo, J.W.; Stappers, L.; Fransaer, J. Stability of sputter-deposited gold nanoparticles in imidazolium ionic liquids. Phys. Chem. Chem. Phys. 2012, 14, 5662–5671. [Google Scholar] [CrossRef] [Green Version]
- Rubim, J.C.; Trindade, F.A.; Gelesky, M.A.; Aroca, R.F.; Dupont, J. Surface-enhanced vibrational spectroscopy of tetrafluoroborate 1-n-butyl-3-methylimidazolium (BMIBF4) ionic liquid on silver surfaces. J. Phys. Chem. C 2008, 112, 19670–19675. [Google Scholar] [CrossRef]
- Guibert, C.; Dupuis, V.; Fresnais, J.; Peyre, V. Controlling nanoparticles dispersion in ionic liquids by tuning the pH. J. Colloid Interface Sci. 2015, 454, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Leon, T.; Jodar-Reyes, A.B.; Bastos-Gonzalez, D.; Ortega-Vinuesa, J.L. Hofmeister effects in the stability and electrophoretic mobility of polystyrene latex particles. J. Phys. Chem. B 2003, 107, 5696–5708. [Google Scholar] [CrossRef] [Green Version]
- Hayes, R.; Warr, G.G.; Atkin, R. At the interface: Solvation and designing ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 1709–1723. [Google Scholar] [CrossRef]
- Canova, F.F.; Matsubara, H.; Mizukami, M.; Kurihara, K.; Shluger, A.L. Shear dynamics of nanoconfined ionic liquids. Phys. Chem. Chem. Phys. 2014, 16, 8247–8256. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.A.; Qiao, R.; Huang, J.S.; Dai, S.; Sumpter, B.G.; Meunier, V. The importance of ion size and electrode curvature on electrical double layers in ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Saringer, S.; Szilagyi, I.; Trefalt, G. A simple method to determine critical coagulation concentration from electrophoretic mobility. Colloids Interfaces 2020, 4, 20. [Google Scholar] [CrossRef]
- Delgado, A.V.; Gonzalez-Caballero, F.; Hunter, R.J.; Koopal, L.K.; Lyklema, J. Measurement and interpretation of electrokinetic phenomena. J. Colloid Interface Sci. 2007, 309, 194–224. [Google Scholar] [CrossRef] [PubMed]
- Mamusa, M.; Siriex-Plenet, J.; Cousin, F.; Dubois, E.; Peyrea, V. Tuning the colloidal stability in ionic liquids by controlling the nanoparticles/liquid interface. Soft Matter 2014, 10, 1097–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SL | SL-IP-2 | |||
|---|---|---|---|---|
| EAN | 0.23 | 7.1 | 0.15 | 15.0 |
| EAN—0.001 M NaNO3 | 0.16 | 8.5 | 0.15 | 12.6 |
| EAN—0.01 M NaNO3 | 0.15 | 7.3 | 0.14 | 16.2 |
| EAN—0.1 M NaNO3 | 0.05 | 7.8 | 0.05 | 14.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takács, D.; Tomšič, M.; Szilagyi, I. Effect of Water and Salt on the Colloidal Stability of Latex Particles in Ionic Liquid Solutions. Colloids Interfaces 2022, 6, 2. https://doi.org/10.3390/colloids6010002
Takács D, Tomšič M, Szilagyi I. Effect of Water and Salt on the Colloidal Stability of Latex Particles in Ionic Liquid Solutions. Colloids and Interfaces. 2022; 6(1):2. https://doi.org/10.3390/colloids6010002
Chicago/Turabian StyleTakács, Dóra, Matija Tomšič, and Istvan Szilagyi. 2022. "Effect of Water and Salt on the Colloidal Stability of Latex Particles in Ionic Liquid Solutions" Colloids and Interfaces 6, no. 1: 2. https://doi.org/10.3390/colloids6010002