Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update

Abstract
:1. Introduction
2. Aims
3. Materials and Methods
4. Results
4.1. Epidemiological Factors
4.2. Preoperative Catecholamine Screening
4.3. Genetic Testing
4.4. Management
5. Discussion
5.1. PGL-1
5.2. PGL-2
5.3. PGL-3
5.4. PGL-4
5.5. PGL-5
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Baysal, B.E. Hereditary paraganglioma targets diverse paraganglia. J. Med. Genet. 2002, 39, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Boedeker, C.C.; Ridder, G.J.; Schipper, J. Paragangliomas of the head and neck: Diagnosis and treatment. Fam. Cancer 2005, 4, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Dziegelewski, P.T.; Knox, A.; Liu, R.; Hung, R.; Harris, J. Familial Paraganglioma Syndrome: Applying Genetic Screening in Otolaryngology. J. Otolaryngol. Head Neck Surg. 2010, 39, 646–653. [Google Scholar]
- Martin, T.P.C.; Irving, R.; Maher, E.R. The genetics of paragangliomas: A review. Clin. Otolaryngol. 2007, 37, 7–11. [Google Scholar]
- Schiavi, F.; Savvoukidis, T.; Trabalzini, F.; Grego, F.; Piazza, M.; Amista, P.; Dematte, S.; Del Piano, A.; Cecchini, M.E.; Erlic, Z.; et al. Paraganglioma syndrome: SDHB, SDHC, and SDHD mutations in head and neck paragangliomas. Ann. N. Y. Acad. Sci. 2006, 1073, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Thabet, M.H.; Kotob, H. Cervical paragangliomas: Diagnosis, management and complications. J. Laryngol. Otol. 2001, 115, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Myssiorek, D. Head and neck paragangliomas: An overview. Otolaryngol. Clin. N. Am. 2001, 34, 829–836. [Google Scholar] [CrossRef]
- Boedeker, C.C. Paragangliomas and paraganglioma syndromes. GMS Curr. Top. Otorhinolaryngol. Head Neck Surg. 2011, 10. [Google Scholar] [CrossRef]
- Badenhop, R.F.; Jansen, J.C.; Fagan, P.A.; Lord, R.S.; Wang, Z.G.; Foster, W.J.; Schofield, P.R. The prevalence of SDHB, SDHC, and SDHD mutations in patients with head and neck paraganglioma and association of mutations with clinical features. J. Med. Genet. 2004, 41, e99. [Google Scholar] [CrossRef] [PubMed]
- Semaan, M.; Megerian, C. Current assessment and management of glomus tumours. Curr. Opin. Otolaryngol. Head Neck Surg. 2008, 16, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Offergeld, C.; Brase, C.; Yaremchuk, S.; Mader, I.; Rischke, H.C.; Glasker, S.; Schmid, K.W.; Wiech, T.; Preuss, S.F.; Suarez, C.; et al. Head and neck paragangliomas: Clinical and molecular genetic classification. Clinics 2012, 67, 19–28. [Google Scholar] [CrossRef]
- Bikhazi, P.H.; Messina, L.; Mhatre, A.N.; Goldstein, J.A.; Lalwani, A.K. Molecular pathogenesis in sporadic head and neck paraganglioma. Laryngoscope 2000, 110, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Barich, F.; Karnell, L.H.; Robinson, R.A.; Zhen, W.K.; Gantz, B.J.; Hoffman, H.T. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer 2002, 94, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Manolidis, S.; Shohet, J.A.; Jackson, C.G.; Glasscock, M.E., 3rd. Malignant glomus tumors. Laryngoscope 1999, 109, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Papaspyrou, K.; Mann, W.J.; Amedee, R.G. Management of head and neck paragangliomas: Review of 120 patients. Head Neck 2009, 31, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.; Kudva, Y.C.; Ebersold, M.J.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A.; Young, W.F., Jr. Benign paragangliomas: Clinical presentation and treatment outcomes in 236 patients. J. Clin. Endocrinol. Metab. 2001, 86, 5210–5216. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.B.; Koeller, K.K.; Adair, C.F. From the archives of the AFIP. Paragangliomas of the head and neck: Radiologic-pathologic correlation. Armed Forces Institute of Pathology. Radiographics 1999, 19, 1605–1632. [Google Scholar] [CrossRef] [PubMed]
- Pellitteri, P.K.; Rinaldo, A.; Myssiorek, D.; Gary Jackson, C.; Bradley, P.J.; Devaney, K.O.; Shaha, A.R.; Netterville, J.L.; Manni, J.J.; Ferlito, A. Paragangliomas of the head and neck. Oral Oncol. 2004, 40, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.B.; Boon, M.S.; Atkins, J.P.; Lowry, L.D. Vagal paraganglioma: The Jefferson experience. Otolaryngol. Head Neck Surg. 2000, 122, 482–487. [Google Scholar] [PubMed]
- Rodriguez-Cuevas, H.; Lau, I.; Rodriguez, H.P. High-altitude paragangliomas diagnostic and therapeutic considerations. Cancer 1986, 57, 672–676. [Google Scholar] [CrossRef]
- Colen, T.Y.; Mihm, F.G.; Mason, T.P.; Roberson, J.B. Catecholamine-secreting paragangliomas: Recent progress in diagnosis and perioperative management. Skull Base 2009, 19, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Osinga, T.E.; Korpershoek, E.; de Krijger, R.R.; Kerstens, M.N.; Dullaart, R.P.; Kema, I.P.; van der Laan, B.F.; van der Horst-Schrivers, A.N.; Links, T.P. Catecholamine-Synthesizing Enzymes Are Expressed in Parasympathetic Head and Neck Paraganglioma Tissue. Neuroendocrinology 2015, 101, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G. Screening for pheochromocytomas and paragangliomas. Curr. Hypertens. Rep. 2012, 14, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, E.; Bresciani, S.; Panaia, R.; Delmastro, E.; Malinverni, G.; Gabriele, P. Hereditary paraganglioma syndrome associated with SDHD gene mutations: A patient with multicentric presentation treated with radiotherapy. Case report. Tumori 2011, 97, 214–220. [Google Scholar] [PubMed]
- Hensen, E.F.; Bayley, J.P. Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam. Cancer 2011, 10, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D. Paragangliomas of the Head and Neck: An Overview from Diagnosis to Genetics. Head Neck Pathol. 2017, 11, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, C.M.; Solorzano, C.C. Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, and Treatment. Surg. Oncol. Clin. N. Am. 2016, 25, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 YEARS OF PARAGANGLIOMA: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [PubMed]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Boedeker, C.C.; Hensen, E.F.; Neumann, H.P.; Maier, W.; van Nederveen, F.H.; Suarez, C.; Kunst, H.P.; Rodrigo, J.P.; Takes, R.P.; Pellitteri, P.K.; et al. Genetics of hereditary head and neck paragangliomas. Head Neck 2014, 36, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Benn, D.E.; Gimenez-Roqueplo, A.P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.; Elston, M.; Gimm, O.; et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.P.; Erlic, Z.; Boedeker, C.C.; Rybicki, L.A.; Robledo, M.; Hermsen, M.; Schiavi, F.; Falcioni, M.; Kwok, P.; Bauters, C.; et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: Cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res. 2009, 69, 3650–3656. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, W.M.; Amdur, R.J.; Vaysberg, M.; Mendenhall, C.M.; Werning, J.W. Head and neck paragangliomas. Head Neck 2011, 33, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kunst, H.P.; Rutten, M.H.; de Monnink, J.P.; Hoefsloot, L.H.; Timmers, H.J.; Marres, H.A.; Jansen, J.C.; Kremer, H.; Bayley, J.P.; Cremers, C.W. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [PubMed]
- Schiavi, F.; Boedeker, C.C.; Bausch, B.; Peczkowska, M.; Gomez, C.F.; Strassburg, T.; Pawlu, C.; Buchta, M.; Salzmann, M.; Hoffmann, M.M.; et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 2005, 294, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Papaspyrou, K.; Mewes, T.; Rossmann, H.; Fottner, C.; Schneider-Raetzke, B.; Bartsch, O.; Schreckenberger, M.; Lackner, K.J.; Amedee, R.G.; Mann, W.J. Head and neck paragangliomas: Report of 175 patients (1989–2010). Head Neck 2012, 34, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar] [PubMed]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Dwight, T.; Benn, D.E.; Clarkson, A.; Vilain, R.; Lipton, L.; Robinson, B.G.; Clifton-Bligh, R.J.; Gill, A.J. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase-deficient gastrointestinal stromal tumors. Am. J. Surg. Pathol. 2013, 37, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Tischler, A.S. Pheochromocytoma and extra-adrenal paraganglioma: Updates. Arch. Pathol. Lab. Med. 2008, 132, 1272–1284. [Google Scholar] [PubMed]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Stoeckli, S.J.; Schuknecht, B.; Alkadhi, H.; Fisch, U. Evaluation of paragangliomas presenting as a cervical mass on color-coded Doppler sonography. Laryngoscope 2002, 112, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R. Imaging and management of head and neck paragangliomas. Eur. Radiol. 2005, 15, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.; Schepers, A.; de Bruine, F.T.; Liauw, L.; Mertens, B.J.; van der Mey, A.G.; van Buchem, M.A. The value of MR angiography techniques in the detection of head and neck paragangliomas. Eur. J. Radiol. 2004, 52, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.; Verbist, B.M.; Mertens, B.J.; van der Mey, A.G.; van Buchem, M.A. Head and neck paragangliomas: Improved tumor detection using contrast-enhanced 3D time-of-flight MR angiography as compared with fat-suppressed MR imaging techniques. AJNR Am. J. Neuroradiol. 2004, 25, 863–870. [Google Scholar] [PubMed]
- Milardovic, R.; Corssmit, E.P.; Stokkel, M. Value of 123I-MIBG Scintigraphy in Paraganglioma. Neuroendocrinology 2010, 91, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hoegerle, S.; Ghanem, N.; Altehoefer, C.; Schipper, J.; Brink, I.; Moser, E.; Neumann, H.P. 18F-DOPA positron emission tomography for the detection of glomus tumours. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, A.; Shulkin, B.L.; Uprimny, C.; Frech, A.; Gasser, R.W.; Url, C.; Gautsch, K.; Madleitner, R.; Nilica, B.; Sprinzl, G.M.; et al. 68Ga-DOTATOC PET/CT provides accurate tumour extent in patients with extraadrenal paraganglioma compared to 123I-MIBG SPECT/CT. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Oldring, D.; Fisch, U. Glomus tumors of the temporal region: Surgical therapy. Am. J. Otol. 1979, 1, 7–18. [Google Scholar] [PubMed]
- Jackson, C.G.; Glasscock, M.E., 3rd; Harris, P.F. Glomus Tumors. Diagnosis, classification, and management of large lesions. Arch. Otolaryngol. 1982, 108, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Fisch, U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann. Otol. Rhinol. Laryngol. 1982, 91, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Alaani, A.; Chavda, S.V.; Irving, R.M. The crucial role of imaging in determining the approach to glomus tympanicum tumours. Eur. Arch. Otorhinolaryngol. 2009, 266, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Shamblin, W.R.; ReMine, W.H.; Sheps, S.G.; Harrison, E.G., Jr. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am. J. Surg. 1971, 122, 732–739. [Google Scholar] [CrossRef]
- Lightowlers, S.; Benedict, S.; Jefferies, S.J.; Jena, R.; Harris, F.; Burton, K.E.; Burnet, N.G. Excellent local control of paraganglioma in the head and neck with fractionated radiotherapy. Clin. Oncol. 2010, 22, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Gilbo, P.; Morris, C.G.; Amdur, R.J.; Werning, J.W.; Dziegielewski, P.T.; Kirwan, J.; Mendenhall, W.M. Radiotherapy for benign head and neck paragangliomas: A 45-year experience. Cancer 2014, 120, 3738–3743. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, W.M.; Amdur, R.J. Radiotherapy for Head and Neck Paraganglioma. Oper. Tech. Otolaryngol. 2016, 27, 55–57. [Google Scholar] [CrossRef]
- Capatina, C.; Ntali, G.; Karavitaki, N.; Grossman, A.B. The management of head-and-neck paragangliomas. Endocr. Relat. Cancer 2013, 20, R291–R305. [Google Scholar] [CrossRef] [PubMed]





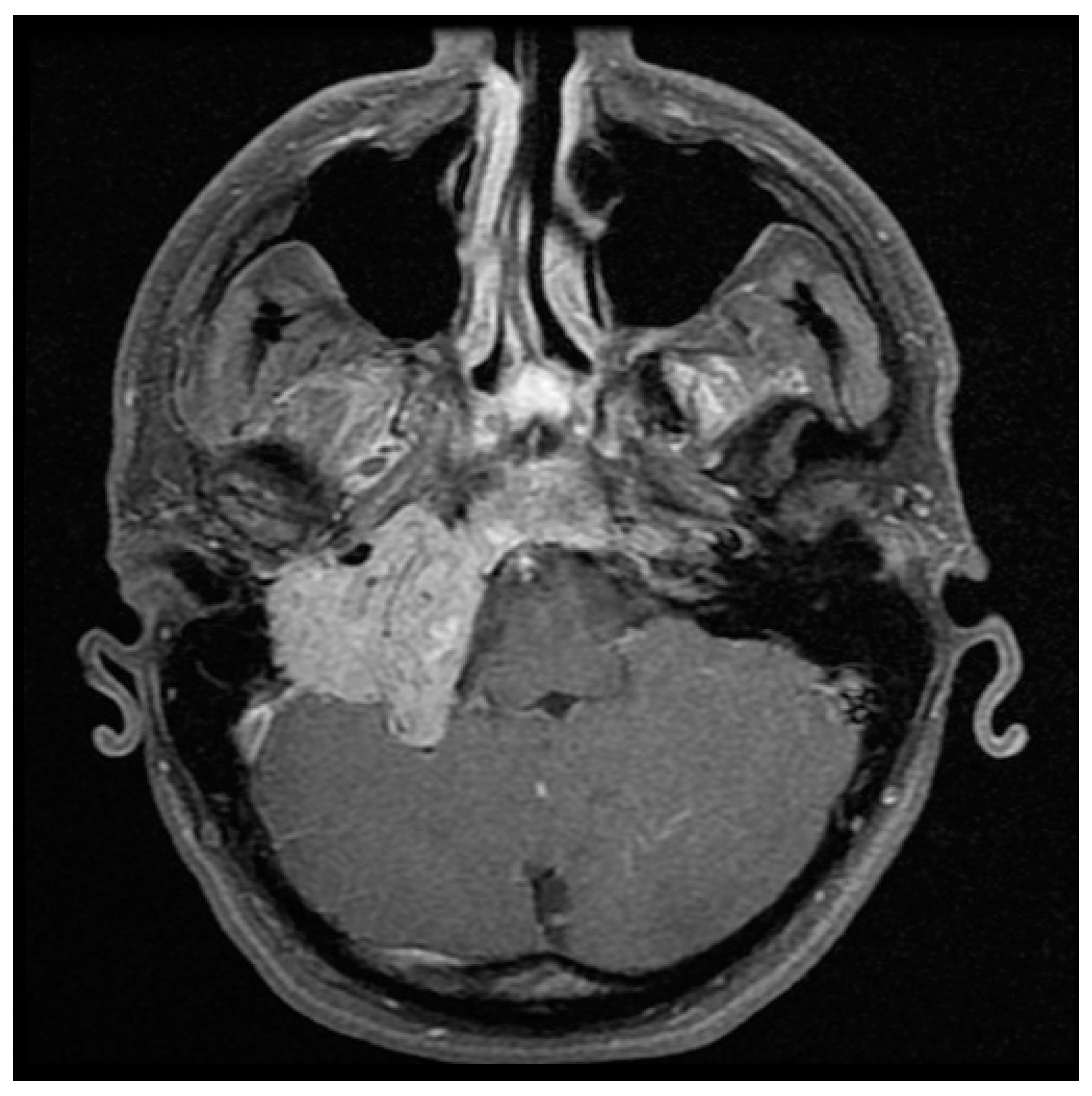{kind=link}
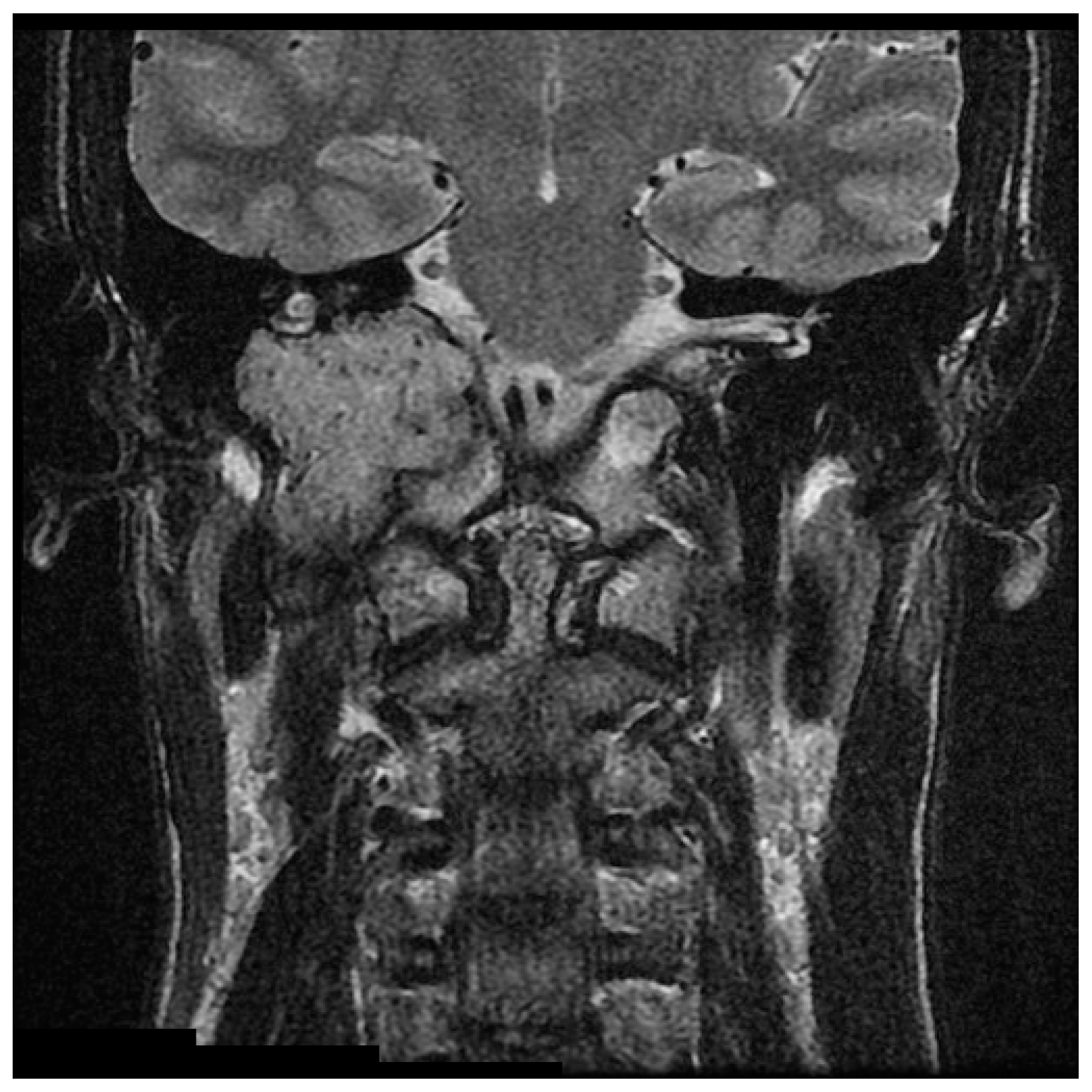{kind=link}
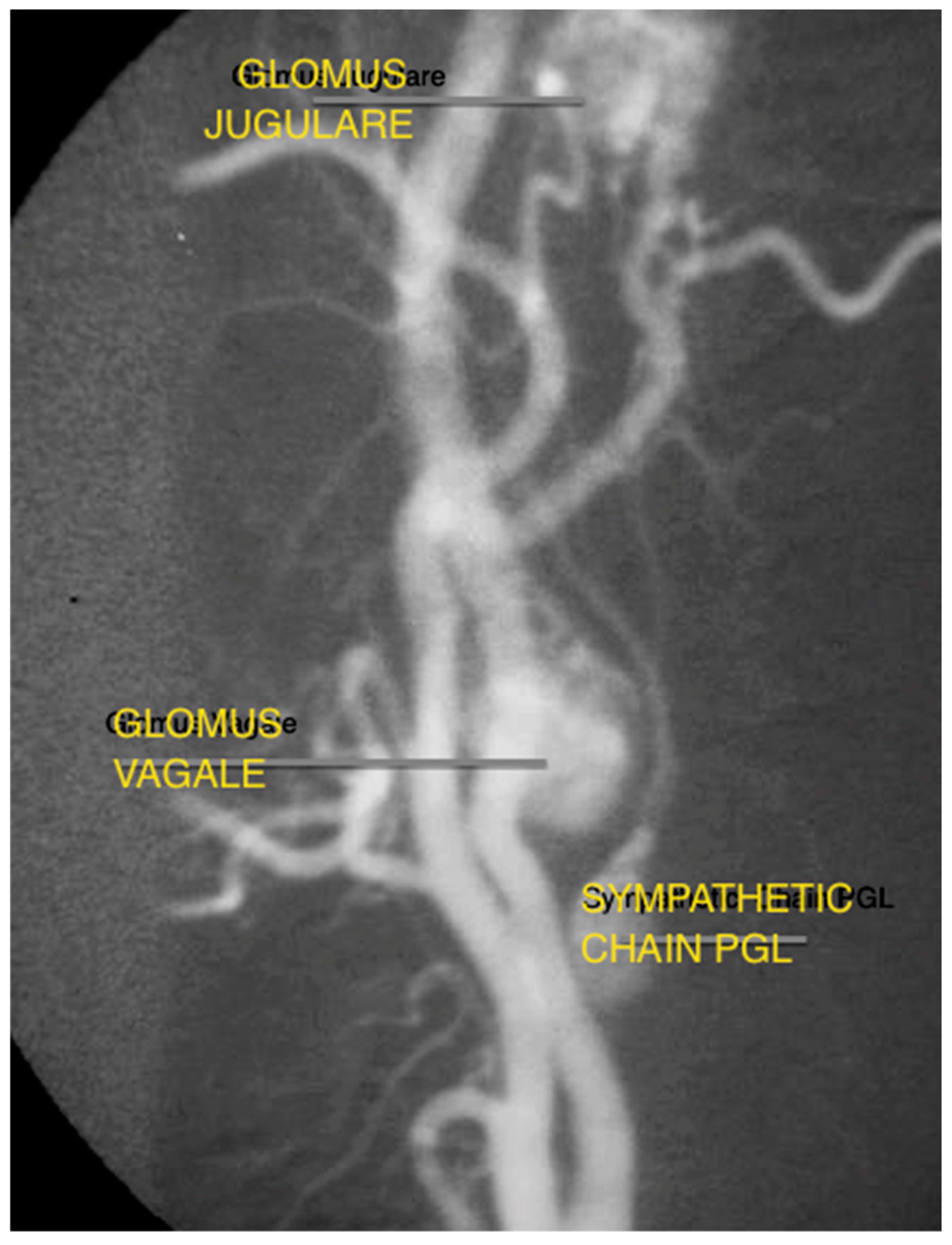{kind=link}
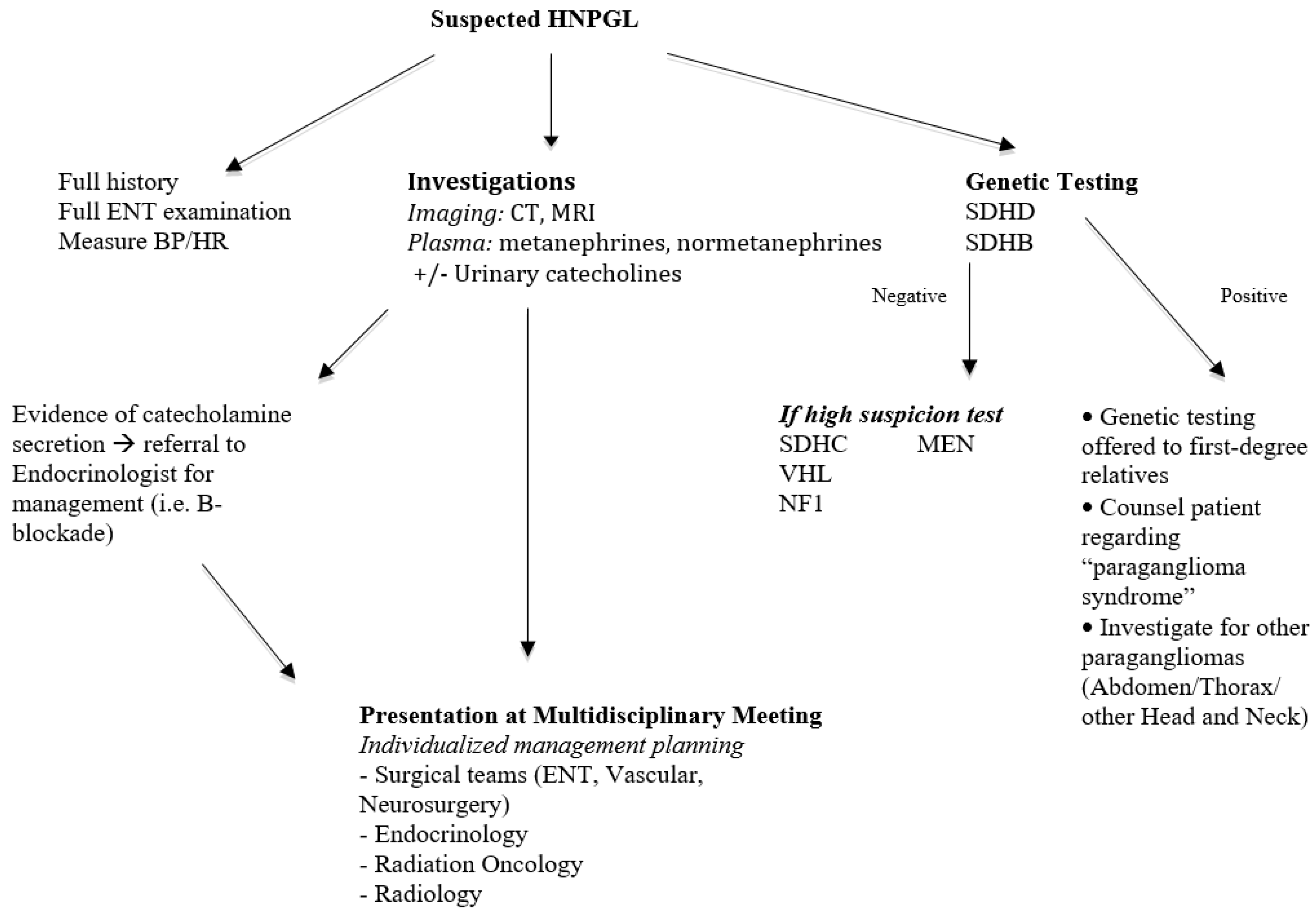{kind=link}
| Glomus Tympanicum | Glomus Jugulare | Glomus Vagale | Carotid Body Tumours | |
|---|---|---|---|---|
| Total Patients (n = 60) | 32 | 20 | 3 | 5 |
| Mean age and Range at presentation (Years) | 52.6 years (30–82 years) | 50.8 years (21–80 years) | 47.0 years (27–57 years) | 38.6 years (30–56 years) |
| Sex differences (Female:Male) | 3:1 | 3:1 | 2:1 | 2:3 |
| Age/Sex (Diagnosis) | Tumour | SDH Mutation | Specific Detail |
|---|---|---|---|
| 34F | Glomus tympanicum | SDHB | Developed extensive recurrence requiring further excision six years post initial surgery |
| 21M | Glomus jugulare | SDHB | Catecholamine secreting tumour. Mother past history phaeochromocytoma. Ongoing elevated catecholamines post planned subtotal excision. |
| 47F | Glomus tympanicum | SDHD | Multiple lesions: contralateral Carotid Body Tumour diagnosed one year post glomus tympanicum excision |
| 31F | Glomus jugulare | SDHD | Monitoring |
| 57F 27F | Glomus vagale Glomus jugulare | SDHB (Presumed SDHB) | Relative (aunt) within pedigree with carotid body tissue |
| 20M | Glomus jugulare | SDHB | Multiple recurrence (four excisions over 20 years, multiple surgeons) |
| 32F | Carotid Body Tumour | SDHD | Multiple lesions: bilateral glomus jugulare concurrently (non-operative management) |
| Glomus Tympanicum (n = 32) | Glomus Jugulare (n = 20) | Glomus Vagale (n = 3) | Carotid Body Tumours (n = 4) |
|---|---|---|---|
| Tympanotomy: 23 | Surgery only: 14 (Infratemporal fossa) | Excision: 2 | Excision: 4 |
| Mastoidectomy: 14 | Five cases combined with Neurosurgery | Observation:1 | |
| Observation: 4 | Radiation only: 4 | ||
| Surgery + Radiotherapy: 5 | |||
| Totals: 41 | Totals: 23 | Totals: 3 | Totals: 4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayward, N.; Cousins, V. Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update. J. Otorhinolaryngol. Hear. Balance Med. 2018, 1, 4. https://doi.org/10.3390/ohbm1010004
Hayward N, Cousins V. Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update. Journal of Otorhinolaryngology, Hearing and Balance Medicine. 2018; 1(1):4. https://doi.org/10.3390/ohbm1010004
Chicago/Turabian StyleHayward, Nathan, and Vincent Cousins. 2018. "Head and Neck Paraganglioma: Medical Assessment, Management, and Literature Update" Journal of Otorhinolaryngology, Hearing and Balance Medicine 1, no. 1: 4. https://doi.org/10.3390/ohbm1010004