
Screening of Potential Indonesia Herbal Compounds Based on Multi-Label Classification for 2019 Coronavirus Disease


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Workflow
- Data preprocessing step, which includes feature extraction on compounds and class data transformation.
- Multilabel modeling step using SAE-DNN model. Hyperparameter tuning is also conducted to find the optimal parameter of SAE-DNN for all feature extraction datasets.
- Post-processing step including model evaluation and herbal compounds prediction.
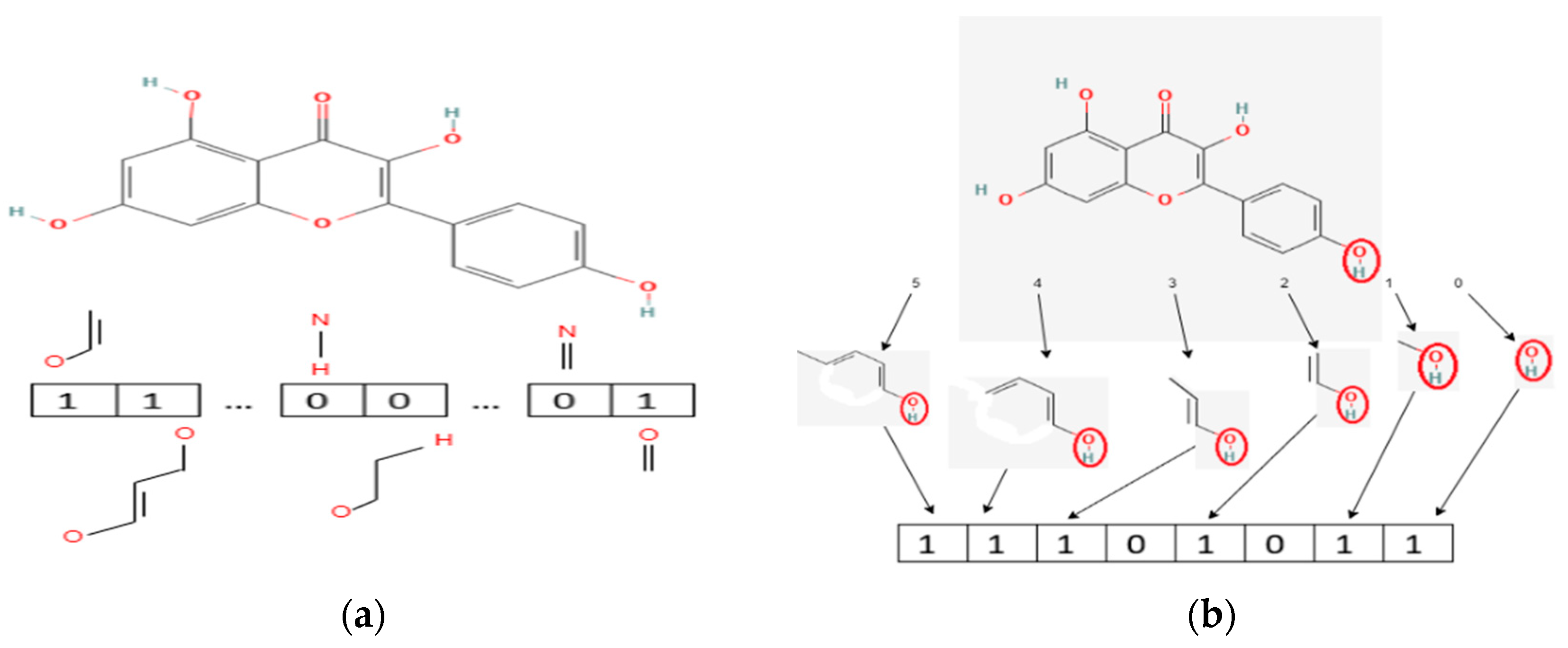
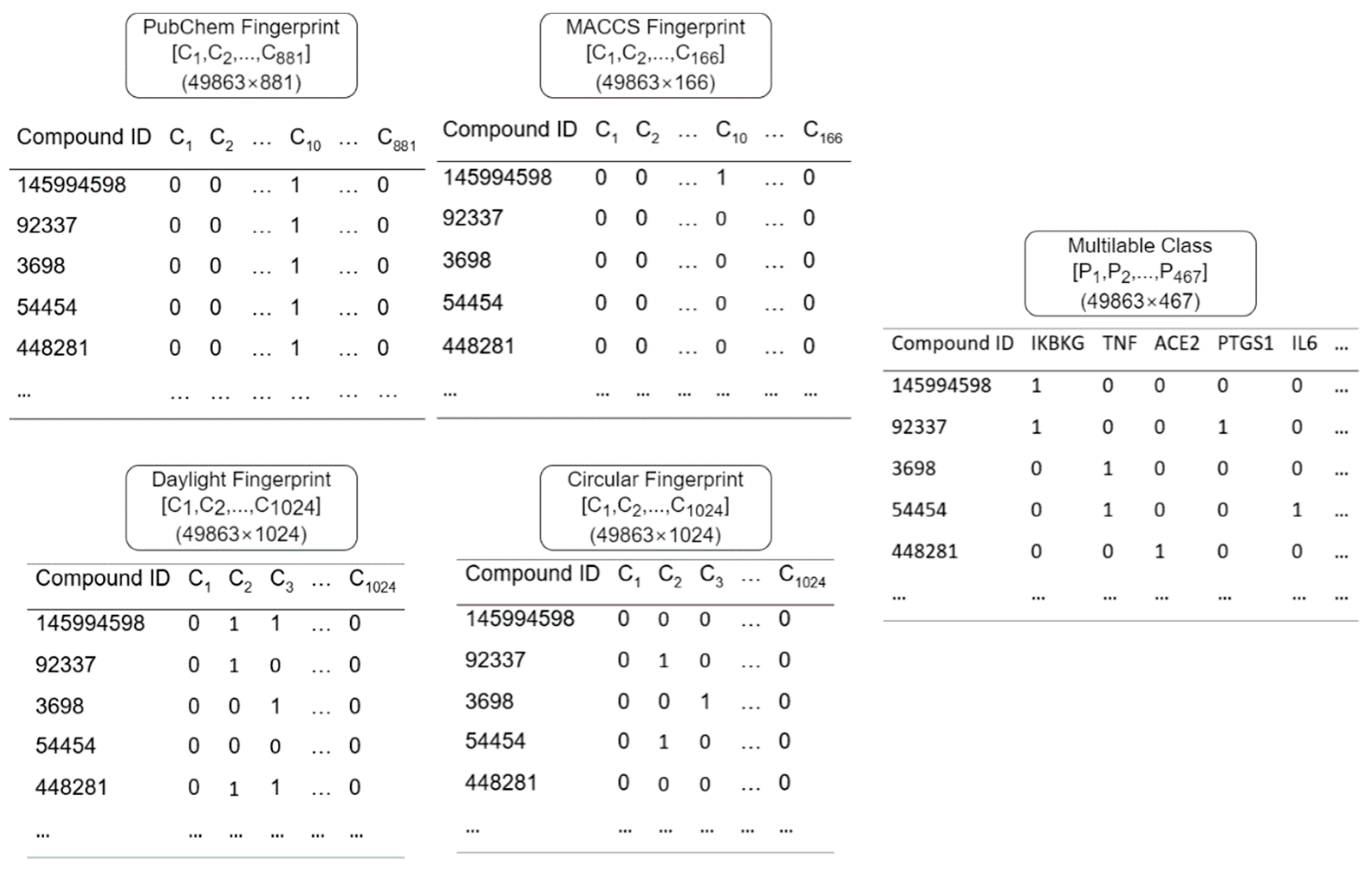
2.3. Data Preprocessing
- Identify each unique compound in the interaction data. There are 49,862 unique compounds in the interaction data
- Identify PubChem ID of each compound
- Identify the SMILES (Simplified Molecular-Input Line-Entry System), which represents the chemical structure of each compound. SMILES data can be seen in Supplementary Spreadsheet 3.
- Form fingerprint of each compound according to the SMILES of each compound.
- The fingerprint feature retrieval process produces a feature vector C (C = [c1, c2, c3, …, cn] with n = the number of substructures on the fingerprint), which will be used as input to the DNN.
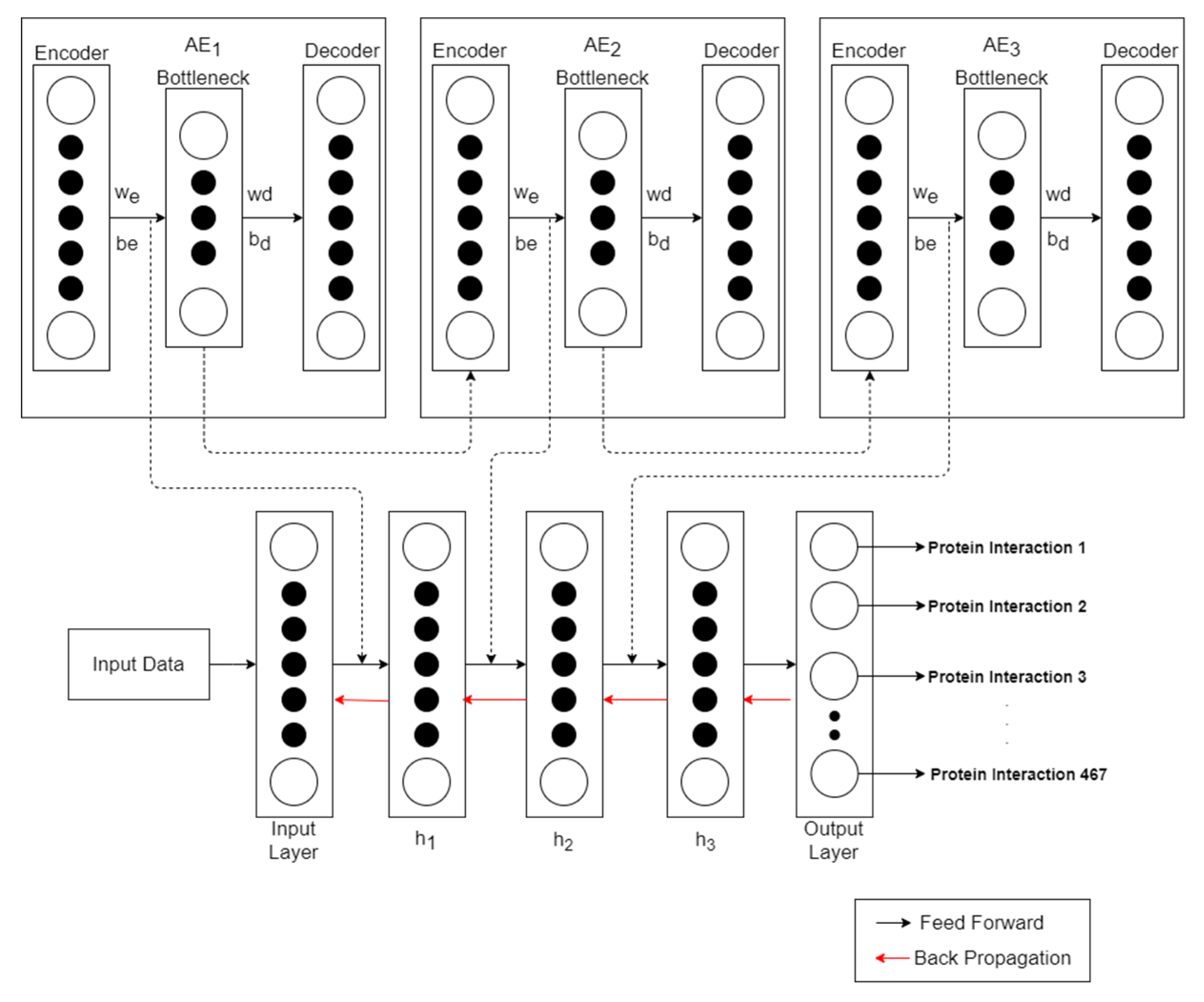
2.4. SAE-DNN Model
| Algorithm 1 pre-training DNN using Stacked AutoEncoder |
| INPUT: Feature vector C |
| OUTPUT: weight (we) and bias (be) for DNN hidden layers |
| 1. Initialize Max Iteration, N as number of AutoEncoder (AE) |
| repeat |
| 2. Train initial AE using C as input |
| for i = 1: N do: |
| 3. Save weight (we) and bias (be) in AE encoder layer |
| 4. Delete AE decoder layer |
| 5. Retrieve data representation at AE bottleneck layer |
| 6. Train next AE using retrieved data representation |
| end |
| until Max Iteration |
| 7. Save weight (we) and bias (be) on all AE encoder layer |
2.5. Postprocess Step
3. Results
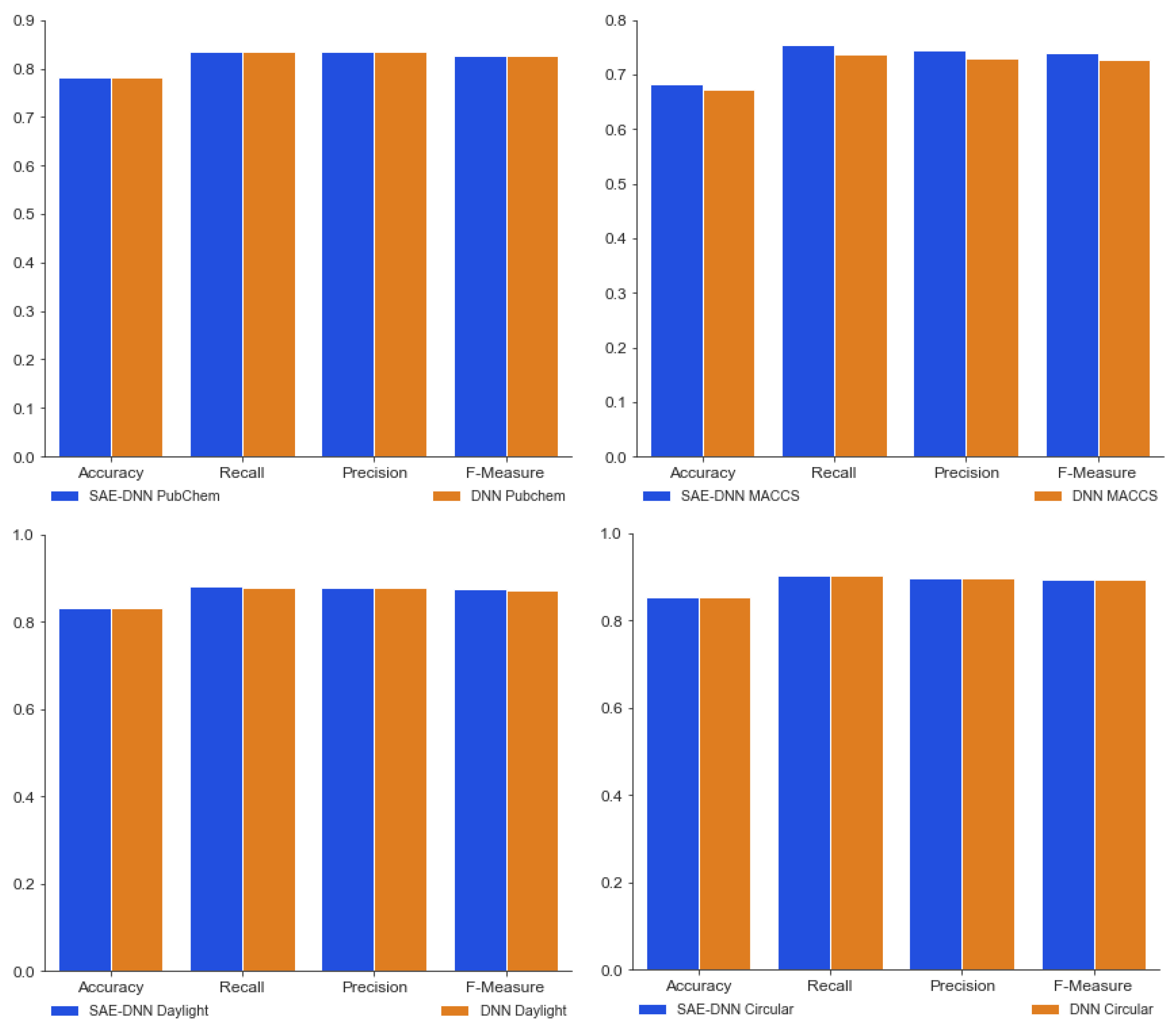
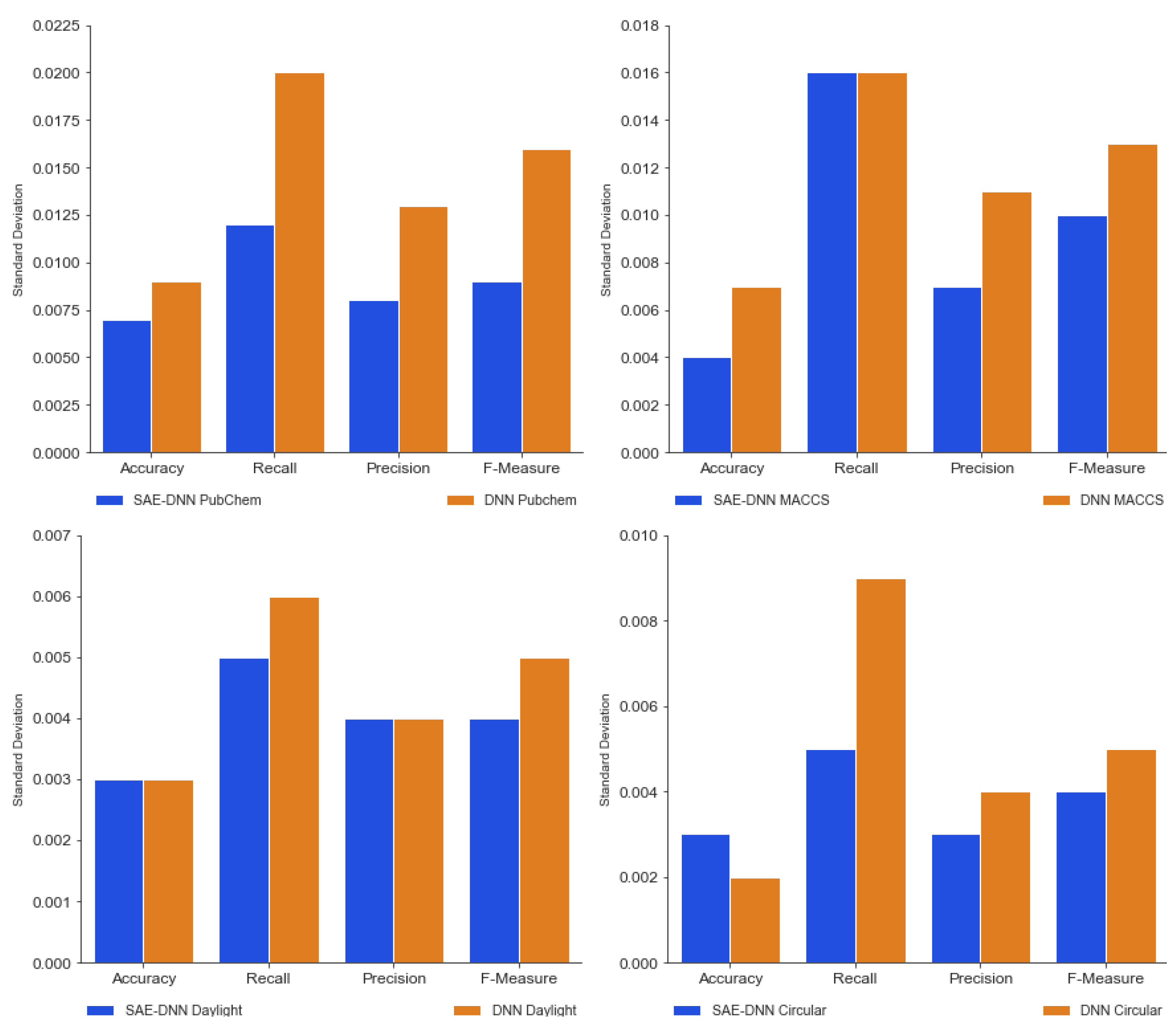
3.1. Performance Comparison between SAE-DNN and DNN Only
3.2. SAE-DNN Performance Comparison for All the Feature Extraction Datasets
3.3. Comparison with Other Approaches from the Literature
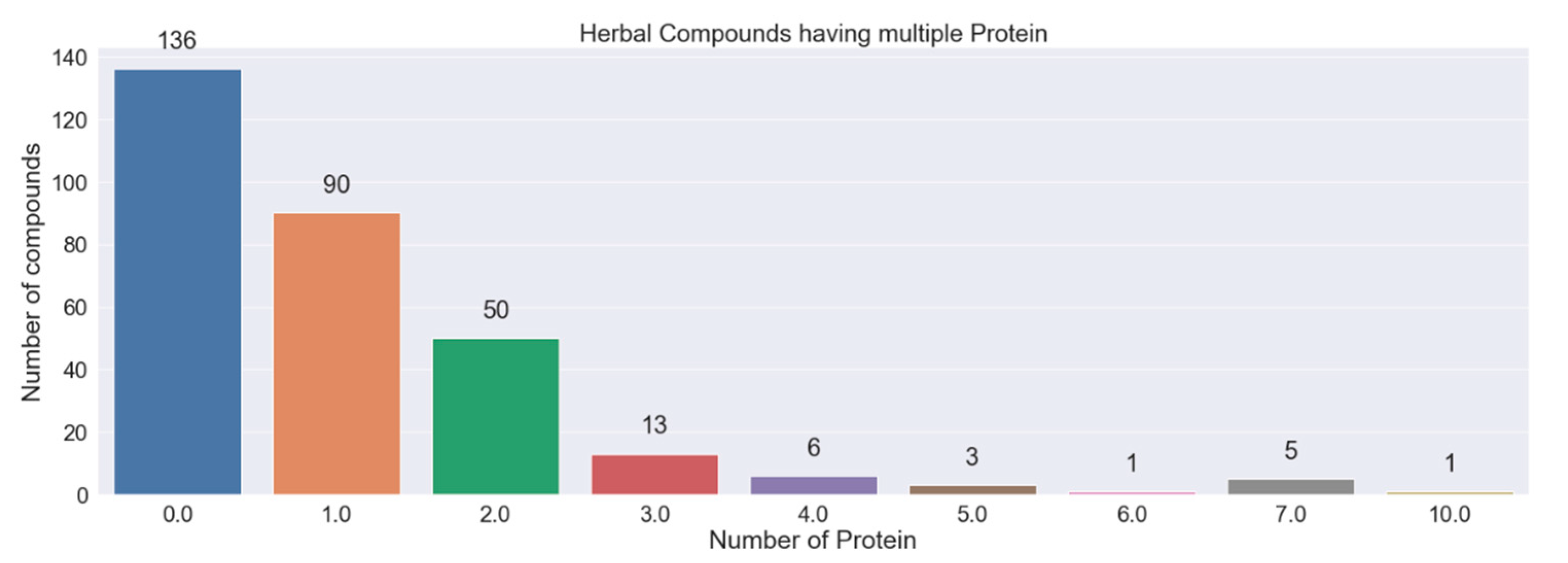
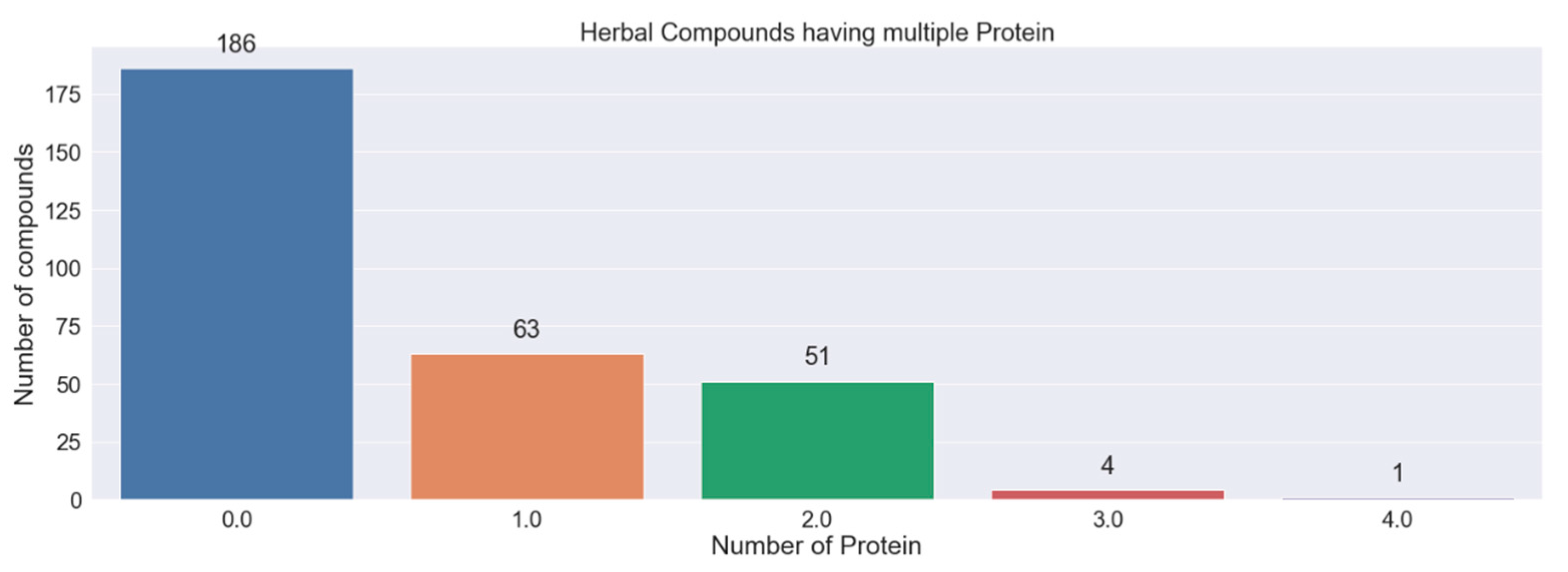
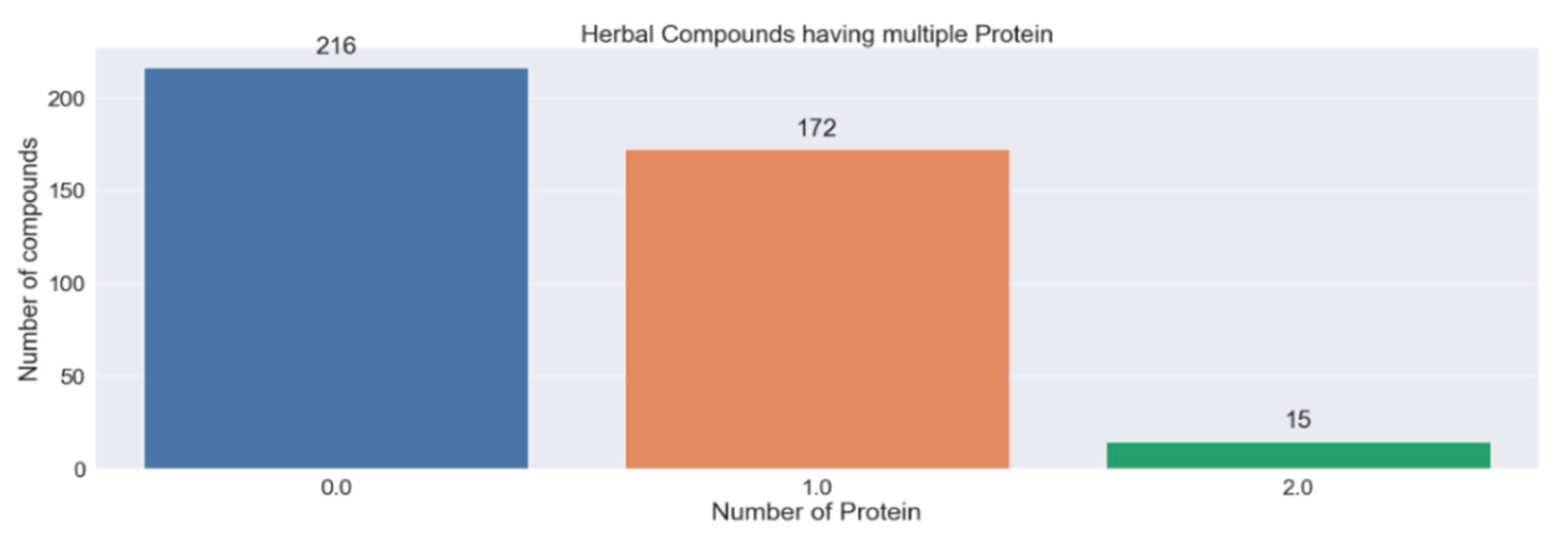
3.4. Herbal Compounds Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Sulistiawan, F.; Kusuma, W.A.; Ramadhanti, N.S.; Tedjo, A. Drug-Target Interaction Prediction in Coronavirus Disease 2019 Case Using Deep Semi-Supervised Learning Model. In Proceedings of the 2020 International Conference on Advanced Computer Science and Information Systems (ICACSIS), Depok, Indonesia, 17–18 October 2020; pp. 83–88. [Google Scholar] [CrossRef]
- Yadav, M.; Dhagat, S.; Eswari, J.S. Emerging Strategies on in Silico Drug Development against COVID-19: Challenges and Opportunities. Eur. J. Pharm. Sci. 2020, 155, 105522. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, C.; Liu, Q.; Zhao, Y.; Zhang, Y.; Qin, Y.; Li, X.; Li, C.; Zhou, C.-Z.; Jin, N.; et al. Identification of Amitriptyline HCl, Flavin Adenine Dinucleotide, Azacitidine and Calcitriol as Repurposing Drugs for Influenza A H5N1 Virus-Induced Lung Injury. PLoS Pathog. 2020, 16, 1–16. [Google Scholar] [CrossRef]
- Erlina, L.; Paramita, R.I.; Kusuma, W.A.; Fadilah, F.; Tedjo, A.; Pratomo, I.P.; Ramadhanti, N.S.; Nasution, A.K.; Surado, F.K.; Fitriawan, A.; et al. Virtual Screening on Indonesian Herbal Compounds as COVID-19 SupportiveTherapy: Machine Learning and Pharmacophore Modeling Approaches. Available online: https://www.researchsquare.com/article/rs-29119/v1 (accessed on 5 November 2021).
- Salim, Z.; Munadi, E. Info Komoditi Tanaman Obat; Badan Pengkajian dan Pengembangan Perdagangan Kementerian Perdagangan Republik Indonesia: Jakarta, Indonesia, 2017. [Google Scholar]
- Ekor, M. The Growing Use of Herbal Medicines: Issues Relating to Adverse Reactions and Challenges in Monitoring Safety. Front. Pharmacol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Larasati, L.; Kusuma, W.A.; Annisa, A. Model Prediksi Interaksi Senyawa Dan Protein Untuk Drug Repositioning Menggunakan Deep Semi-Supervised Learning. J. Teknol. Inf. Dan Ilmu Komput. 2020, 7, 727. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A Network Integration Approach for Drug-Target Interaction Prediction and Computational Drug Repositioning from Heterogeneous Information. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.A.K.; Lin, H.H.; Chen, Y.Z.; Li, Z.R.; Cao, Z. Efficacy of Different Protein Descriptors in Predicting Protein Functional Families. BMC Bioinform. 2007, 8, 300. [Google Scholar] [CrossRef] [Green Version]
- Fernández-De Gortari, E.; García-Jacas, C.R.; Martinez-Mayorga, K.; Medina-Franco, J.L. Database Fingerprint (DFP): An Approach to Represent Molecular Databases. J. Cheminform. 2017, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.I.; Keum, J.; Nam, H.I. DeepConv-DTI: Prediction of Drug-Target Interactions via Deep Learning with Convolution on Protein Sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef] [Green Version]
- Zagidullin, B.; Wang, Z.; Guan, Y.; Pitkänen, E.; Tang, J. Comparative Analysis of Molecular Fingerprints in Prediction of Drug Combination Effects. Brief. Bioinform. 2021, 22, bbab291. [Google Scholar] [CrossRef]
- Mei, S.; Zhang, K. A Multi-Label Learning Framework for Drug Repurposing. Pharmaceutics 2019, 11, 466. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, S.M.H.; Chen, W.; Meng, H.; Jahan, H.; Liu, Y.; Hasan, S.M.M. Prediction of Drug-Target Interaction Based on Protein Features Using Undersampling and Feature Selection Techniques with Boosting. Anal. Biochem. 2020, 589, 113507. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Shan, X.; Chen, T.; Jiang, M.; Wang, Y.; Wang, Q.; Salahub, D.R.; Xiong, Y.; Wei, D.Q. DTI-MLCD: Predicting Drug-Target Interactions Using Multi-Label Learning with Community Detection Method. Brief. Bioinform. 2021, 22, bbaa205. [Google Scholar] [CrossRef]
- Pliakos, K.; Vens, C.; Tsoumakas, G. Predicting Drug-Target Interactions With Multi-Label Classification and Label Partitioning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 1596–1607. [Google Scholar] [CrossRef]
- Fitriawan, A.; Wasito, I.; Syafiandini, A.F.; Amien, M.; Yanuar, A. Multi-Label Classification Using Deep Belief Networks for Virtual Screening of Multi-Target Drug. In Proceedings of the 2016 International Conference on Computer, Control, Informatics and its Applications (IC3INA): Recent Progress in Computer, Control, and Informatics for Data Science, Tangerang, Indonesia, 3–5 October 2016; pp. 102–107. [Google Scholar] [CrossRef]
- Pliakos, K.; Vens, C. Drug-Target Interaction Prediction with Tree-Ensemble Learning and Output Space Reconstruction. BMC Bioinform. 2020, 21, 1V. [Google Scholar] [CrossRef] [PubMed]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of Drug–Target Interaction Networks from the Integration of Chemical and Genomic Spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, A.; Li, R.; Yang, B.; Weng, H.; Ou, A.; Hong, H.; Zhou, Z.; Gong, P.; Zhang, C. Deep Learning Architectures for Multi-Label Classification of Intelligent Health Risk Prediction. BMC Bioinform. 2017, 18, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK Family Databases: Integrated Metabolite-Plant Species Databases for Multifaceted Plant Research. Plant Cell Physiol. 2011, 53, e1. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 2016, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for Exploring Drug-Target Relationships. Nucleic Acids Res. 2008, 36, D919–D922. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A Comprehensive Resource for in Silico Drug Discovery and Exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Yanuar, A.; Mun’im, A.; Bertha, A.; Lagho, A.; Syahdi, R.R.; Rahmat, M.; Suhartanto, H. Medicinal Plants Database and Three Dimensional Structure of the Chemical Compounds from Medicinal Plants in Indonesia. arXiv 2011, arXiv:1111.7183. [Google Scholar]
- PubChem Substructure Fingerprint V1.3. Available online: https://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt (accessed on 14 July 2021).
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daylight Fingerprints-Screening and Similarity. Available online: https://www.daylight.com/dayhtml/doc/theory/theory.finger.html (accessed on 15 July 2021).
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Modeling 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES. 2. Algorithm for Generation of Unique SMILES Notation. J. Chem. Inf. Comput. Sci. 2002, 29, 97–101. [Google Scholar] [CrossRef]
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular Fingerprint Similarity Search in Virtual Screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef]
- Bahi, M.; Batouche, M. Drug-Target Interaction Prediction in Drug Repositioning Based on Deep Semi-Supervised Learning. IFIP Adv. Inf. Commun. Technol. 2018, 522, 302–313. [Google Scholar] [CrossRef]
- Ioffe, S.; Szegedy, C. Batch Normalization: Accelerating Deep Network Training by Reducing Internal Covariate Shift. Available online: http://proceedings.mlr.press/v37/ioffe15.pdf (accessed on 27 August 2021).
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Salakhutdinov, R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar] [CrossRef]
- Chen, G.; Chen, P.; Shi, Y.; Hsieh, C.-Y.; Liao, B.; Zhang, S. Rethinking the Usage of Batch Normalization and Dropout in the Training of Deep Neural Networks. arXiv 2019, arXiv:1905.05928. [Google Scholar]
- Shahriari, B.; Swersky, K.; Wang, Z.; Adams, R.P.; de Freitas, N. Taking the Human out of the Loop: A Review of Bayesian Optimization. Proc. IEEE 2016, 104, 148–175. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, T.; Bursztein, E.; Long, J.; Chollet, F.; Jin, H.; Invernizzi, L. Others Keras Tuner 2019. Available online: https://github.com/keras-team/keras-tuner (accessed on 27 August 2021).
- Szymański, P.; Kajdanowicz, T. A Network Perspective on Stratification of Multi-Label Data. In Proceedings of the First International Workshop on Learning with Imbalanced Domains: Theory and Applications; 2017. Available online: http://proceedings.mlr.press/v74/szyma%C5%84ski17a.html (accessed on 27 August 2021).
- Sechidis, K.; Tsoumakas, G.; Vlahavas, I. On the Stratification of Multi-Label Data. In Lecture Notes in Computer Science (Including Subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics) LNAI; 2011; Volume 6913, pp. 145–158. Available online: http://lpis.csd.auth.gr/publications/sechidis-ecmlpkdd-2011.pdf (accessed on 27 August 2021). [CrossRef] [Green Version]
- Boulila, W.; Driss, M.; Al-Sarem, M.; Saeed, F.; Krichen, M. Weight Initialization Techniques for Deep Learning Algorithms in Remote Sensing: Recent Trends and Future Perspectives. arXiv 2021, arXiv:2102.07004. [Google Scholar]
- Charles, Z.; Papailiopoulos, D. Stability and Generalization of Learning Algorithms That Converge to Global Optima. Available online: http://proceedings.mlr.press/v80/charles18a/charles18a.pdf (accessed on 19 November 2021).
- El-Behery, H.; Attia, A.F.; El-Feshawy, N.; Torkey, H. Efficient Machine Learning Model for Predicting Drug-Target Interactions with Case Study for Covid-19. Comput. Biol. Chem. 2021, 93, 107536. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Stokes, J.M.; Eastman, R.T.; Itkin, Z.; Zakharov, A.V.; Collins, J.J.; Jaakkola, T.S.; Barzilay, R. Deep Learning Identifies Synergistic Drug Combinations for Treating COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118, e2105070118. [Google Scholar] [CrossRef]
- Hao, M.; Bryant, S.H.; Wang, Y. A New Chemoinformatics Approach with Improved Strategies for Effective Predictions of Potential Drugs. J. Cheminform. 2018, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Wang, D.W.; Chen, C. The Potential Effects of DPP-4 Inhibitors on Cardiovascular System in COVID-19 Patients. J. Cell. Mol. Med. 2020, 24, 10274. [Google Scholar] [CrossRef]
- Zhang, F.; Mears, J.R.; Shakib, L.; Beynor, J.I.; Shanaj, S.; Korsunsky, I.; Nathan, A.; Accelerating Medicines Partnership Rheumatoid Arthritis and Systemic Lupus Erythematosus (AMP RA/SLE) Consortium; Donlin, L.T.; Raychaudhuri, S. IFN-γ and TNF-α Drive a CXCL10+ CCL2+ Macrophage Phenotype Expanded in Severe COVID-19 Lungs and Inflammatory Diseases with Tissue Inflammation. Genome Med. 2021, 13, 64. [Google Scholar] [CrossRef]
- Aboudounya, M.M.; Heads, R.J. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediat. Inflamm. 2021, 2021, 8874339. [Google Scholar] [CrossRef]
- Vagapova, E.R.; Lebedev, T.D.; Prassolov, V.S. Viral Fibrotic Scoring and Drug Screen Based on MAPK Activity Uncovers EGFR as a Key Regulator of COVID-19 Fibrosis. Sci. Rep. 2021, 11, 11234. [Google Scholar] [CrossRef] [PubMed]
- Wambier, C.G.; Goren, A.; Vaño-Galván, S.; Ramos, P.M.; Ossimetha, A.; Nau, G.; Herrera, S.; McCoy, J. Androgen Sensitivity Gateway to COVID-19 Disease Severity. Drug Dev. Res. 2020, 81, 771–776. [Google Scholar] [CrossRef]
- Kim, S.-J.; Um, J.-Y.; Hong, S.-H.; Lee, J.-Y. Anti-Inflammatory Activity of Hyperoside through the Suppression of Nuclear Factor-ΚB Activation in Mouse Peritoneal Macrophages. Am. J. Chin. Med. 2011, 39, 171–181. [Google Scholar] [CrossRef]
- Ma, Y.; Tang, T.; Sheng, L.; Wang, Z.; Tao, H.; Zhang, Q.; Zhang, Y.; Qi, Z. Aloin Suppresses Lipopolysaccharide-Induced Inflammation by Inhibiting JAK1-STAT1/3 Activation and ROS Production in RAW264.7 Cells. Int. J. Mol. Med. 2018, 42, 1925–1934. [Google Scholar] [CrossRef] [Green Version]
- Park, M.-Y.; Kwon, H.-J.; Sung, M.-K. Evaluation of Aloin and Aloe-Emodin as Anti-Inflammatory Agents in Aloe by Using Murine Macrophages. Biosci. Biotechnol. Biochem. 2009, 73, 828–832. [Google Scholar] [CrossRef]
- Santo, B.L.S.D.E.; Santana, L.F.; Junior, W.H.K.; Araújo, F.D.O.D.; Bogo, D.; Freitas, K.D.C.; Guimarães, R.D.C.A.; Hiane, P.A.; Pott, A.; Filiú, W.F.D.O.; et al. Medicinal Potential of Garcinia Species and Their Compounds. Molecules 2020, 25, 4513. [Google Scholar] [CrossRef]
- Jnawali, H.N.; Lee, E.; Jeong, K.-W.; Shin, A.; Heo, Y.-S.; Kim, Y. Anti-Inflammatory Activity of Rhamnetin and a Model of Its Binding to c-Jun NH 2-Terminal Kinase 1 and P38 MAPK. J. Nat. Prod. 2014, 77, 258–263. [Google Scholar] [CrossRef]
- Xing, X.; Wang, H. Anti-Asthmatic Effect of Laurotetanine Extracted from Litsea Cubeba (Lour.) Pers. Root on Ovalbumin-Induced Allergic Asthma Rats, and Elucidation of Its Mechanism of Action. Trop. J. Pharm. Res. 2021, 18, 1277–1283. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Sadeghipour, P.; Aghakouchakzadeh, M.; Kakavand, H.; Ariannejad, H.; Connors, J.M.; Hunt, B.J.; Berger, J.S.; van Tassell, B.W.; Middeldorp, S.; et al. Use of Novel Antithrombotic Agents for COVID-19: Systematic Summary of Ongoing Randomized Controlled Trials. J. Thromb. Haemost. 2021, 19, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; von Mering, C.; Campillos, M.; Jensen, L.J.; Bork, P. STITCH: Interaction Networks of Chemicals and Proteins. Nucleic Acids Res. 2008, 36, D684–D688. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyperparameter | Values |
|---|---|
| HL0 Node | 100–2000 |
| HLi Node | 0.5 × (HL0)–0.75 × (HL0) |
| Hidden layer | 1–6 |
| Optimizer | Adam, adagrad |
| Learning rate | 0.01–0.1 |
| Dropout rate | 0.2–0.7 |
| Hyperparameter | Model | |||
|---|---|---|---|---|
| PubChem | Daylight | MACCS | Circular | |
| HL0 Node | 1500 | 2000 | 1024 | 2000 |
| HLi Node | 0.5 | 0.5 | 0.5 | 0.75 |
| Hidden layer | 2 | 2 | 3 | 2 |
| Optimizer | Adam | Adam | Adam | Adam |
| Learning rate | 0.01 | 0.01 | 0.01 | 0.01 |
| Dropout rate | 0.5 | 0.5 | 0.5 | 0.5 |
| Metrics | Model | |||
|---|---|---|---|---|
| PubChem | Daylight | MACCS | Circular | |
| Accuracy | 0.78747 ± 0.005 | 0.82814 ± 0.004 | 0.68272 ± 0.004 | 0.83160 ± 0.007 |
| Recall | 0.86178 ± 0.004 | 0.89306 ± 0.012 | 0.75462 ± 0.016 | 0.91836 ± 0.005 |
| Precision | 0.84641 ± 0.003 | 0.87854 ± 0.004 | 0.74407 ± 0.007 | 0.88848 ± 0.005 |
| F-measure | 0.84572 ± 0.003 | 0.87808 ± 0.006 | 0.74089 ± 0.010 | 0.89368 ± 0.005 |
| Compounds | Proteins | Probability | Protein Relevance Score | Total Herbal Plants | Commonly Used Herbal Plants |
|---|---|---|---|---|---|
| Rutin | DPP4, PTGS1 | 0.534, 0.628 | 12.445, 0.895 | 1 | Carmellia sinensis (tea leaves) |
| Damnacanthal | TNF | 0.819 | 12.213 | 2 | Morinda citrifolia L. (noni) |
| Ascorbic acid | F3, TLR4, FURIN, PPT1, PVR, PPARA, FADS2 | 0.998, 0.999, 0.909, 0.994, 0.863, 0.937, 0.969 | 11.928, 10.370, 4.519, 0.965, 0.965, 0.895, 0.653 | 5 | Mangifera indica (mango) Carica papaya (papaya) |
| Palmitoleic acid | F3, TLR4, FURIN, PPT1, PVR, PPARA, FADS2 | 0.997, 0.999, 0.961, 0.994, 0.915, 0.932, 0.982 | 11.928, 10.370, 4.519, 0.965, 0.965, 0.895, 0.653 | 4 | Mangifera indica (mango) Punica granatum (pomegranate) |
| Petunidin | F3, TLR4, FURIN, PPT1, PVR, PPARA, FADS2 | 0.997, 0.999, 0.961, 0.994, 0.915, 0.932, 0.982 | 11.928, 10.370, 4.519, 0.965, 0.965, 0.895, 0.653 | 1 | Lagerstroemia indica (crepe-myrtle) |
| Naringin | F3, TLR4, FURIN, PPT1, PVR, PPARA, FADS2 | 0.997, 0.999, 0.961, 0.994, 0.915, 0.932, 0.982 | 11.928, 10.370, 4.519, 0.965, 0.965, 0.895, 0.653 | 4 | Punica granatum (pomegranate) Citrus aurantium (bitter orange) |
| Malvidin | F3, TLR4, PPT1, PPARA | 0.996, 0.931, 0.883, 0.832 | 11.928, 10.370, 0.965, 0.895 | 3 | Impatiens balsamina Melastoma malabathricum |
| Sterculic acid | F3, TLR4, PPT1, PPARA | 0.996, 0.931, 0.883, 0.832 | 11.928, 10.370, 0.965, 0.895 | 2 | Cassia fistula Sterculia foetida |
| Ricinoleic acid | F3, TLR4, FADS2, PPARA | 0.975, 0.999, 0.999, 0.715 | 11.928, 10.370, 0.965, 0.895 | 2 | Ganoderma lucidum Ricinus communis (ricinus) |
| P-coumaric acid | F3, FADS2, PPARA, PPT1, ELOVL5 | 0.983, 0.99, 0.983, 1.0, 0.999 | 10.370, 0.965, 0.895, 0.653, 0.653 | 22 | Mangifera indica (mango) Punica granatum (pomegranate) |
| Compounds | Proteins | Probability | Protein Relevance Score | Total Herbal Plants | Commonly Used Herbal Plants |
|---|---|---|---|---|---|
| Hyperoside | EGFR | 0.509 | 9.887 | 7 | Mangifera indica (mango) |
| Safrole | TNFRSF1A | 0.586 | 5.459 | 1 | Cananga odorata |
| Estradiol | TNFRSF1A, CSNK2B, EIF3F | 0.528, 0.923, 0.626 | 5.459, 0.965, 0.653 | 1 | Punica granatum (pomegranate) |
| Tetrahydroxyflavone | TNFRSF1A, ALOX5 | 0.788, 0.537 | 5.459, 0.895 | 5 | Cucumis sativus (cucumber) |
| Myristic acid | TNFRSF1A, ALOX5, EIF3F | 0.912, 0.509, 0.608 | 5.459, 0.895, 0.653 | 16 | Mangifera indica (mango) |
| Rhamnetin | TNFRSF1A, ALOX5, EIF3F | 0.916, 0.526, 0.603 | 5.459, 0.895, 0.653 | 6 | Averrhoa carambola (starfruit) |
| A-terpinene | TTR | 0.564 | 2.638 | 20 | Cuminum cyminum L. (white cumin) |
| Epicatechin | AR | 0.820 | 2.428 | 19 | Punica granatum (pomegranat3) |
| Proanthocyanidin a2 | AR | 0.627 | 2.428 | 5 | Garcinia mangostana (mangosteen) |
| Momordicilin | AR | 0.813 | 2.428 | 1 | Momordica charantia |
| Compounds | Proteins | Probability | Protein Relevance Score | Total Herbal Plants | Commonly Used Herbal Plants |
|---|---|---|---|---|---|
| Glucobrassicin | EGFR | 0.986 | 9.887 | 8 | Raphanus sativus (radish) Brassica oleracea (wild cabbage) |
| Cuminaldehyde | EGFR | 0.923 | 9.887 | 4 | Cuminum cyminum L. (cumin) Eucalyptus globulus |
| P-cymene | EGFR | 0.904 | 9.887 | 23 | Mangifera indica (mango) Nigella sativa (black cumin) |
| Methyl cinnamate | EGFR | 0.895 | 9.887 | 1 | Ocimum (basil) |
| Garcimangosone d | EGFR | 0.856 | 9.887 | 1 | Garcinia mangostana (mangosteen) |
| Ethyl cinnamate | EGFR | 0.85 | 9.887 | 1 | Durio zibethinus (durian) |
| Kaempferol | EGFR | 0.844 | 9.887 | 1 | Carthamus tinctorius (safflower) |
| Cinnamic acid | EGFR | 0.817 | 9.887 | 3 | Glycine max (soybean) Ocimum basilicum (basil) |
| P-coumaric acid | EGFR | 0.808 | 9.887 | 22 | Mangifera indica (mango) Punica granatum (pomegranate) |
| Cinnamaldehyde | EGFR | 0.801 | 9.887 | 4 | Carica papaya (papaya) Pogostemon cablin (patchouli) |
| Compounds | Protein Predicted by SAE-DNN Model | Species | Activity | References | ||
|---|---|---|---|---|---|---|
| Circular | Daylight | PubChem | ||||
| Hyperoside | AHR, AKT1 | EGFR | PRKCA | Mangifera indica (mango) | Served as an anti-inflammatory | [52] |
| Aloin | MBL2 | LGALS3 | PRKCA | Aloe vera | Indicate to induce anti-inflammatory | [53,54] |
| Garcimangosone d | RELA, NFKB1 | RELA, NFKB1 | EGFR | Garcinia mangostana (mangosteen) | Garcinia mangostana can be used to cure inflammation | [55] |
| Rhamnetin | CSNK2B, ALOX5, EIF3F | TNFRSF1A, ALOX5, EIF3F | PRKCA | Averrhoa carambola (starfruit) | Have good anti-inflammatory activity | [56] |
| Anisaldehyde | TXNRD1, PLOD1, P4HA1, TFRC, PLOD3, PLOD2 | PLOD1, P4HA1, PLOD3, PLOD2 | EGFR | Pimpinella anisum | - | - |
| Laurotetanine | ACHE | ADRB2 | PRKCA | Litsea cubeba | Possess anti-inflammatory properties | [57] |
| Momordin i | PTGS1, TOP1 | TOP1 | PRKCA | Basella rubra L. | - | - |
| Isoquercetin | RAC1 | RAC1 | PRKCA | Mangifera indica (mango) | Have anti-inflammatory properties | [58] |
| Cycloeucalenone | TOP1 | TOP1 | CSNK2A1 | Musa sapientum | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadli, A.; Kusuma, W.A.; Annisa; Batubara, I.; Heryanto, R. Screening of Potential Indonesia Herbal Compounds Based on Multi-Label Classification for 2019 Coronavirus Disease. Big Data Cogn. Comput. 2021, 5, 75. https://doi.org/10.3390/bdcc5040075
Fadli A, Kusuma WA, Annisa, Batubara I, Heryanto R. Screening of Potential Indonesia Herbal Compounds Based on Multi-Label Classification for 2019 Coronavirus Disease. Big Data and Cognitive Computing. 2021; 5(4):75. https://doi.org/10.3390/bdcc5040075
Chicago/Turabian StyleFadli, Aulia, Wisnu Ananta Kusuma, Annisa, Irmanida Batubara, and Rudi Heryanto. 2021. "Screening of Potential Indonesia Herbal Compounds Based on Multi-Label Classification for 2019 Coronavirus Disease" Big Data and Cognitive Computing 5, no. 4: 75. https://doi.org/10.3390/bdcc5040075