
Multi-Omics Data Reveal Amino Acids Related Genes in the Common Carp Cyprinus carpio
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. The Determination of Amino Acids Content
2.3. Single Nucleotide Polymorphism Genotyping
2.4. Genome-Wide Association Analysis
2.5. Transcriptome Sequencing and Differential Expression Analysis
2.6. DNA Methylation Sequencing and Differential Methylation Analysis
3. Results
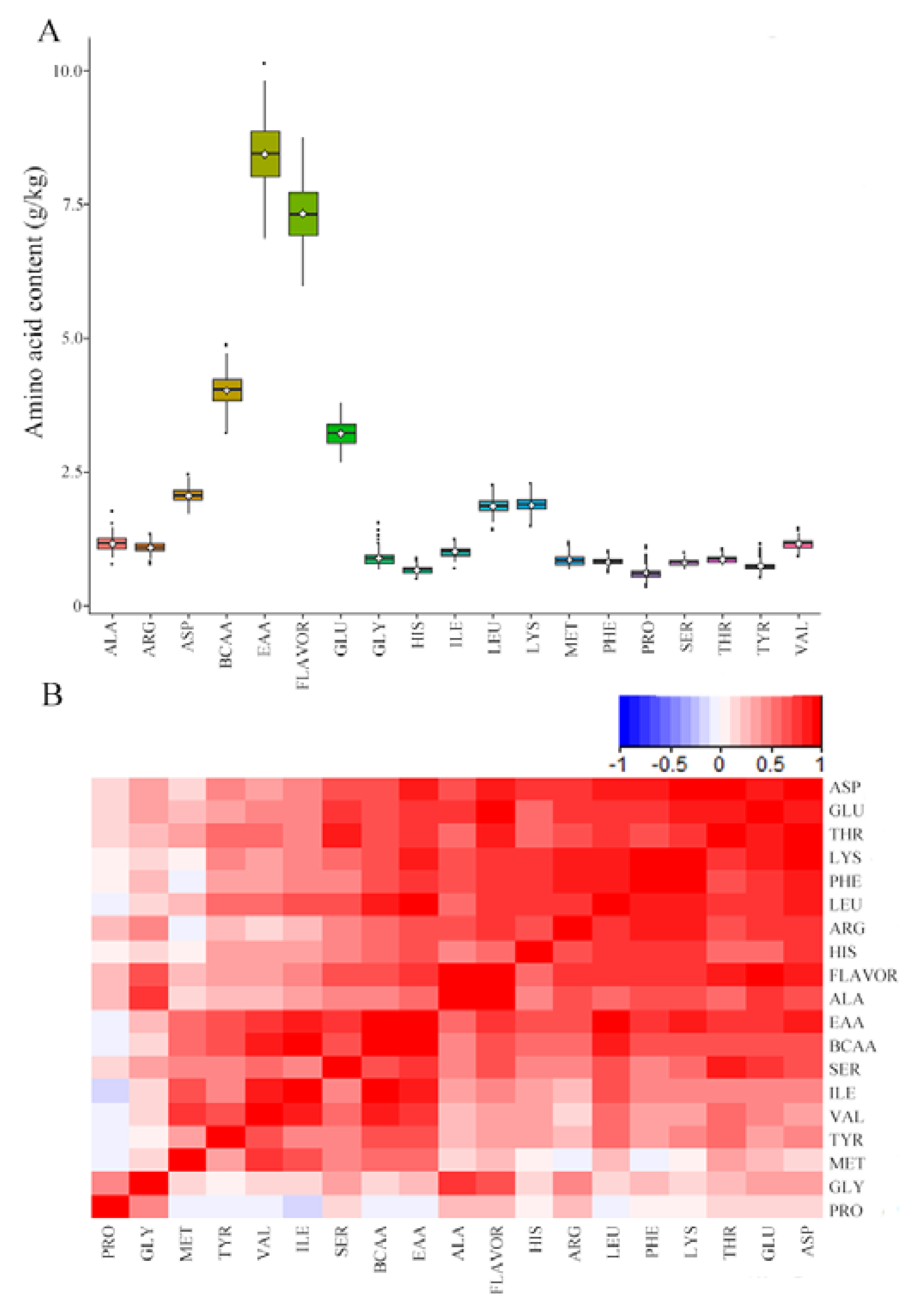
3.1. Genotyping and Phenotyping of C. carpio Samples
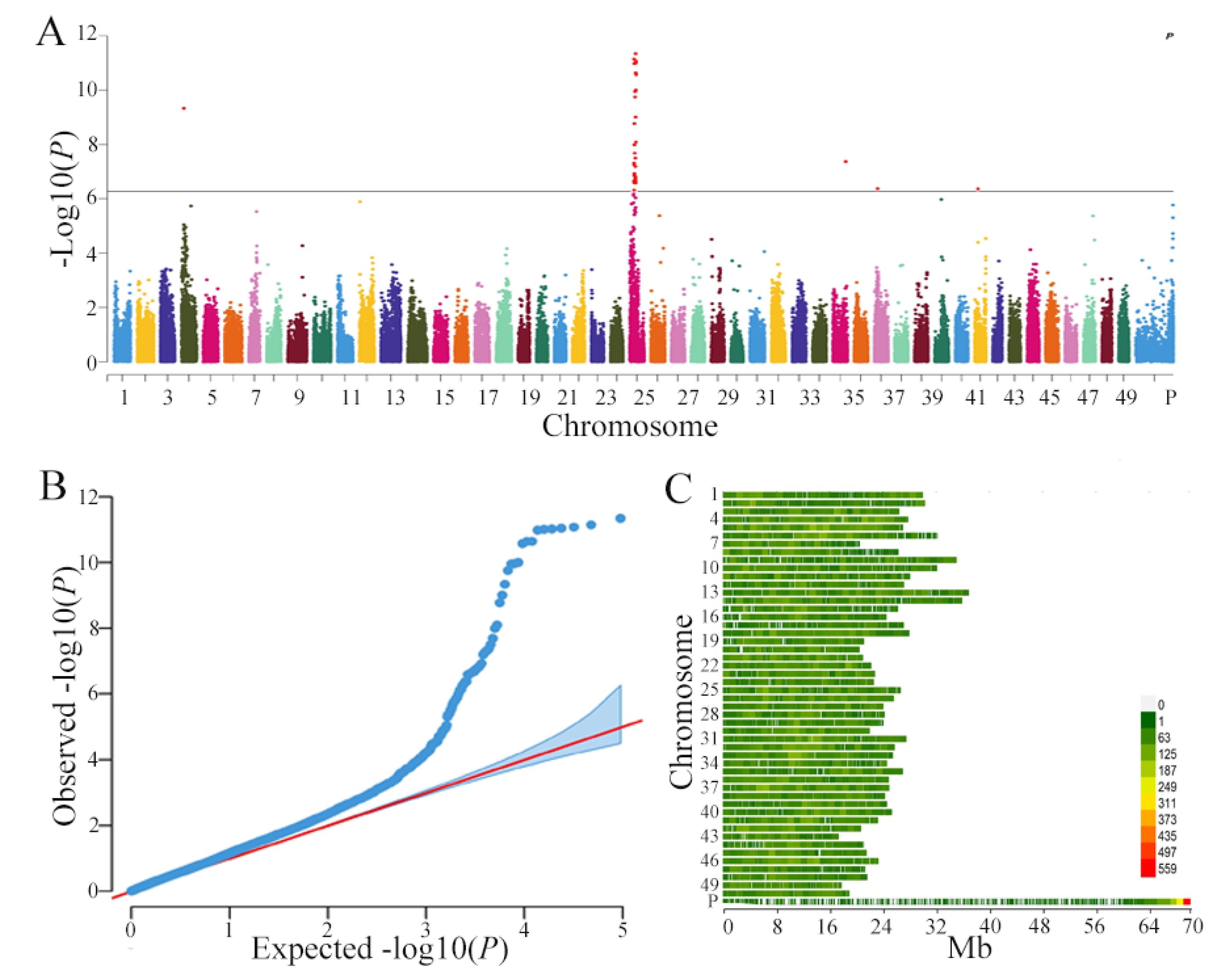
3.2. Genome-Wide Association Analysis
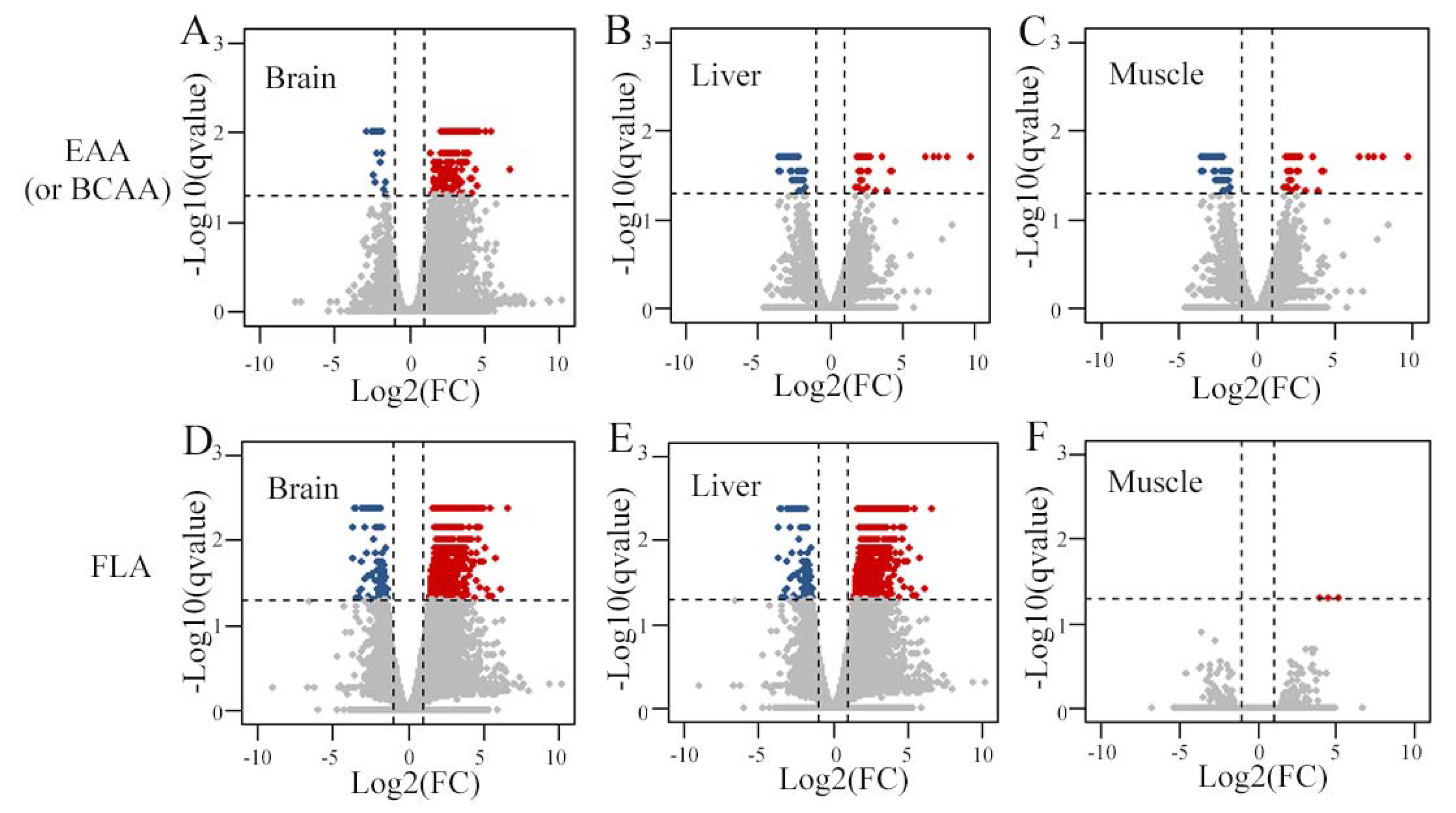
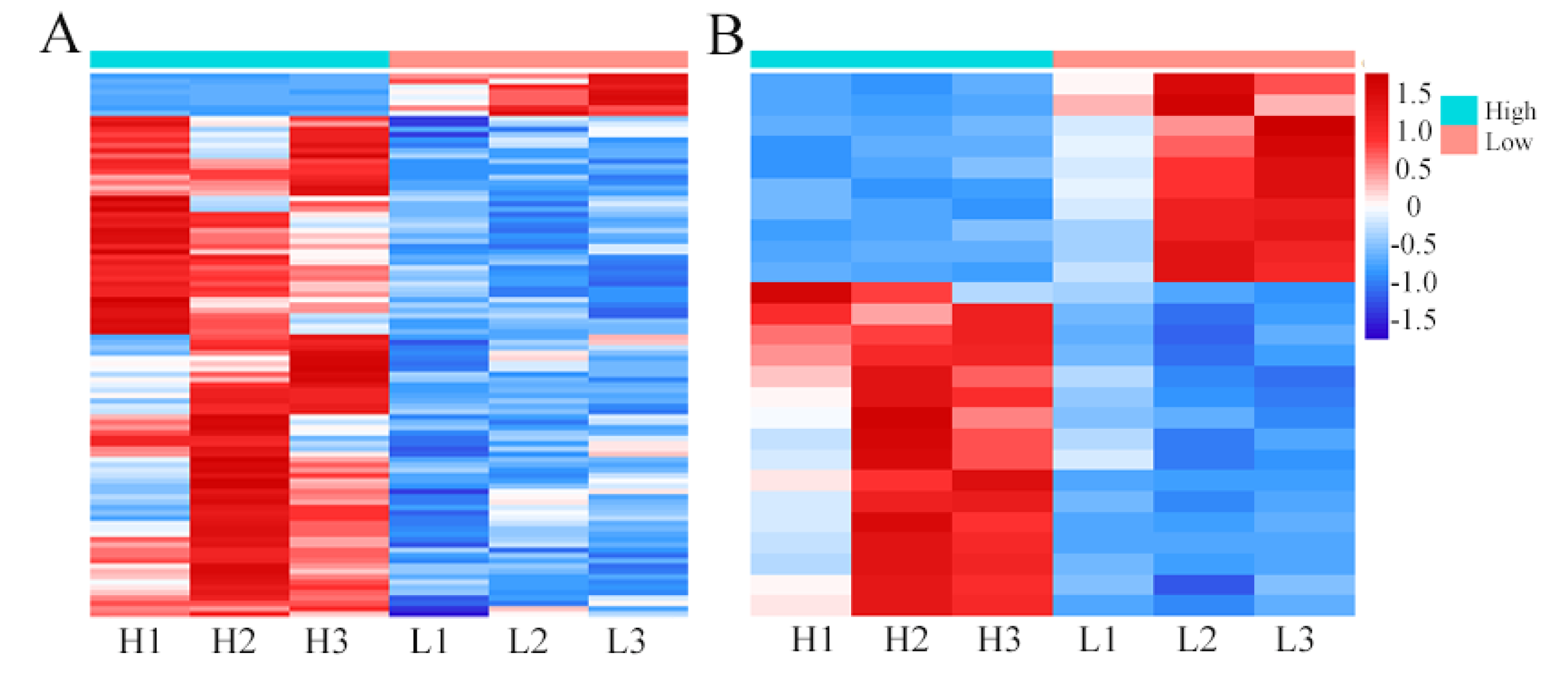
3.3. Transcriptomic Analysis of Divergent Amino Acid Content
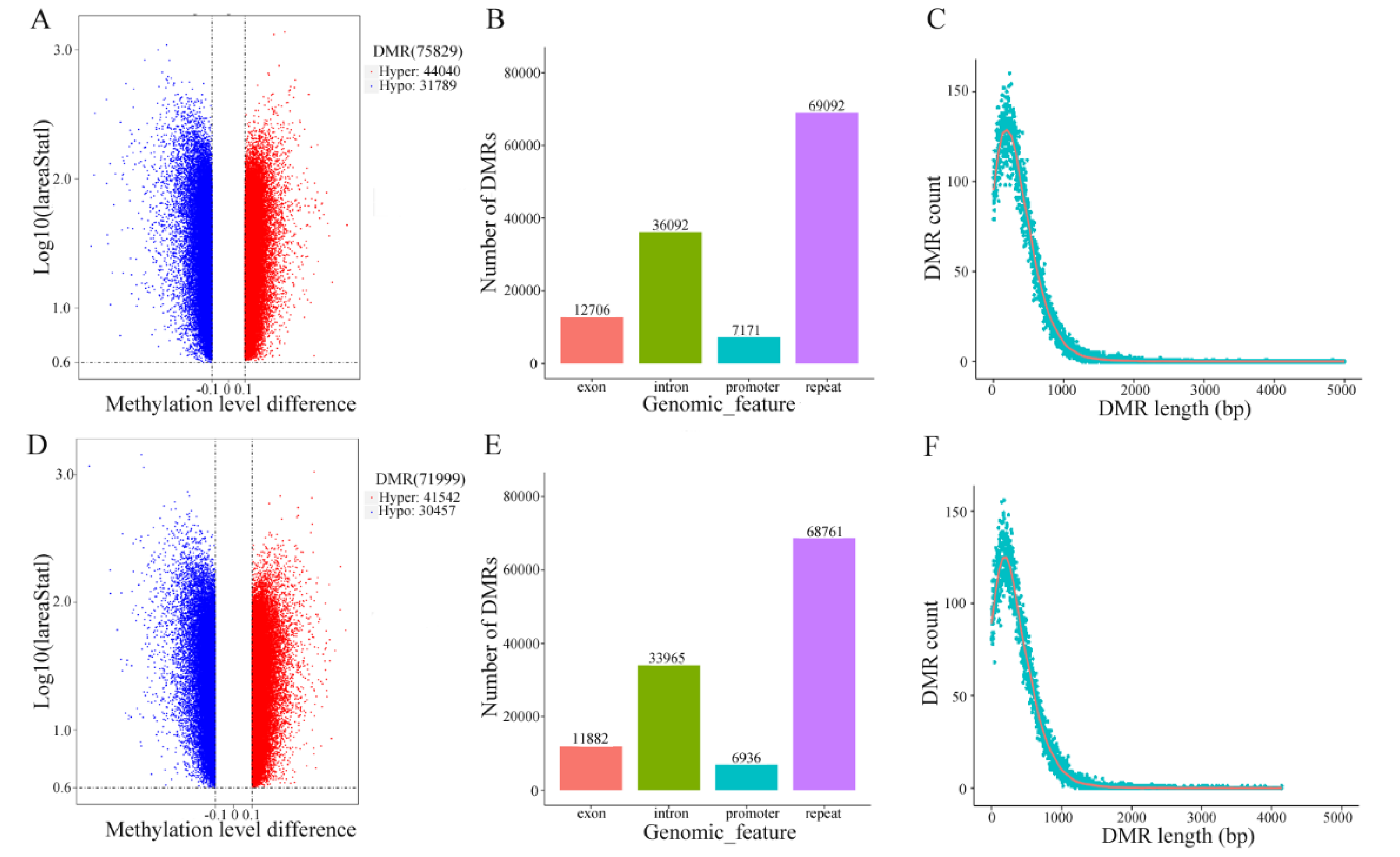
3.4. Differential Methylation Analysis
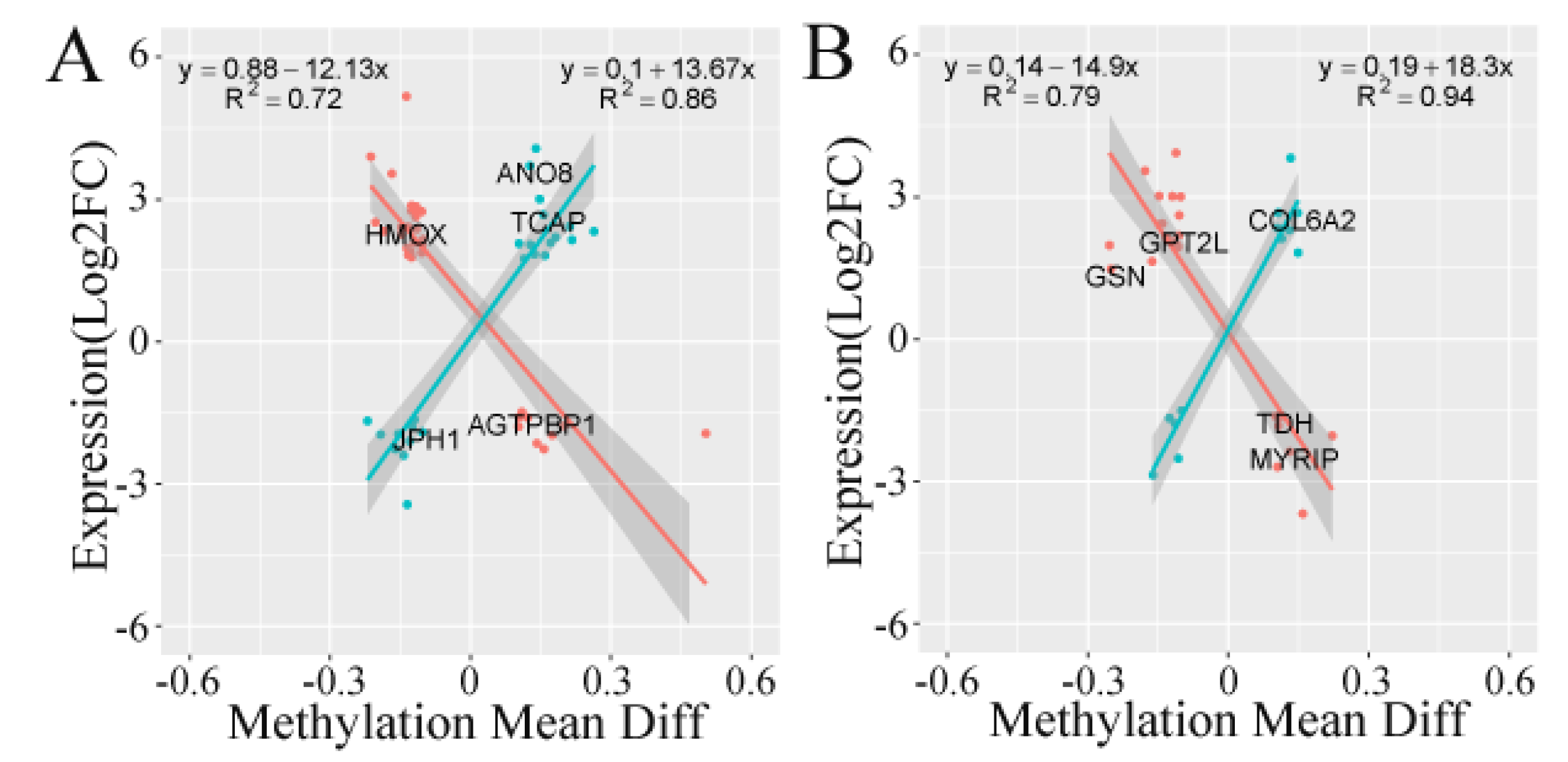
3.5. Correlation Analysis of Transcriptome and Methylation Results
4. Discussion
4.1. Genes Associated with Muscular Amino Acid Content by GWAS
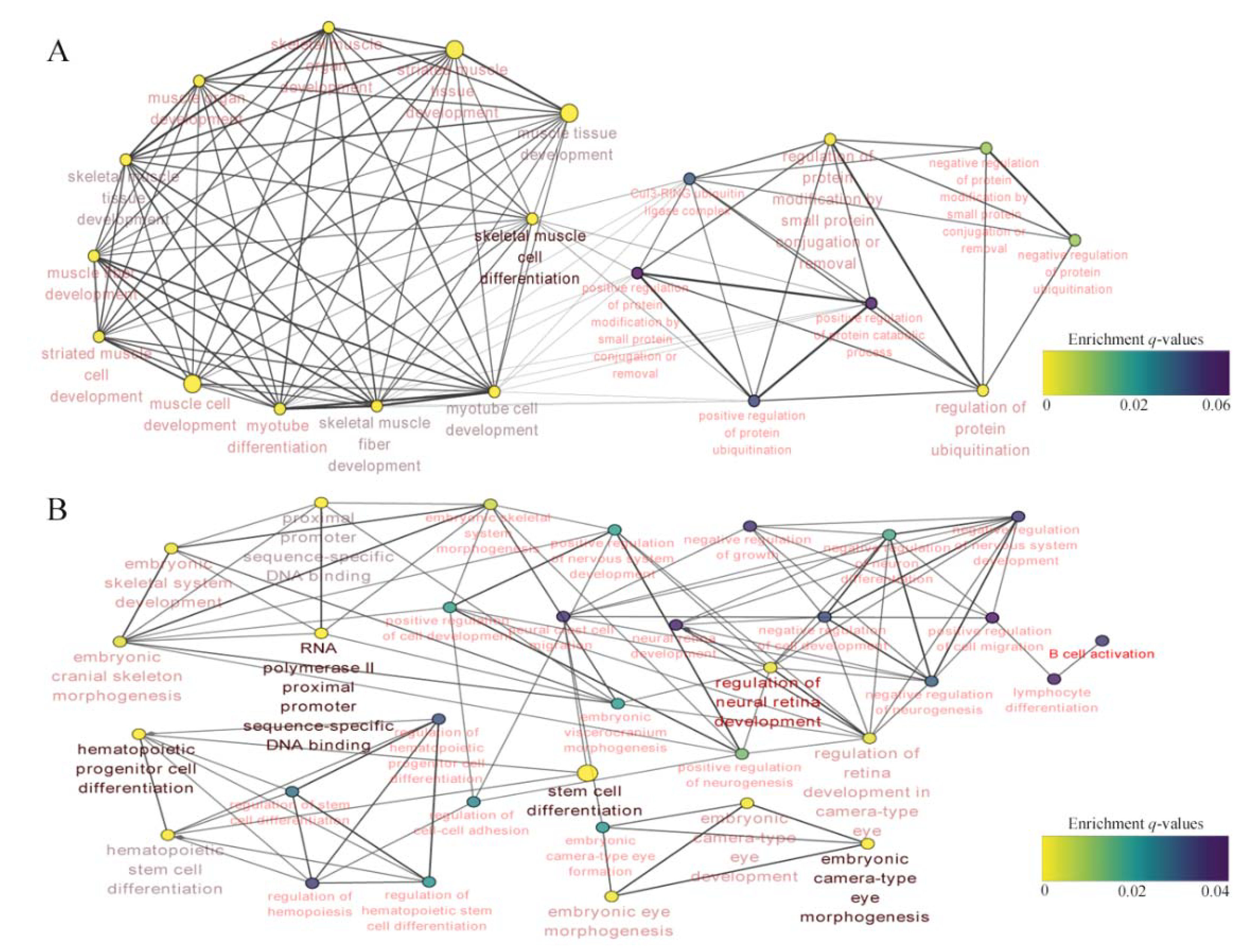
4.2. Network Analysis Based on RNA-Seq Data
4.3. Gene Identification with DMP and Integration Incorporating DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EAA | essential amino acid; |
| NEAA | nonessential amino acid; |
| FAA | functional amino acid; |
| FLA | flavorous amino acid; |
| BCAA | branched-chain amino acids; |
| GWAS | genome-wide association study; |
| WGBS | whole-genome bisulfite sequencing; |
| GO | gene ontology; |
| KEGG | Kyoto encyclopedia of genes and genomes; |
| DEG | differentially expressed gene; |
| DMP | differentially methylated promoter; |
| DMR | differentially methylated region |
References
- FAO. Fishery and Aquaculture Statistics; Food and Agriculture Organization: Rome, Italy, 2017. [Google Scholar]
- Xu, J.; Zhao, Z.; Zhang, X.; Zheng, X.; Li, J.; Jiang, Y.; Kuang, Y.; Zhang, Y.; Feng, J.; Li, C.; et al. Development and evaluation of the first high-throughput SNP array for common carp (Cyprinus carpio). BMC Genom. 2014, 15, 307. [Google Scholar]
- Peng, W.; Jian, X.; Yan, Z.; Feng, J.; Peng, X. An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio). Sci. Rep. 2016, 6, 26693. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, P.; Jiang, Y.; Zhao, Z.; Feng, J.; Tai, R.; Dong, C.; Xu, J. Genomic, transcriptomic, and epigenomic features differentiate genes that are relevant for muscular polyunsaturated fatty acids in the common carp. Front. Genet. 2019, 10, 217. [Google Scholar] [PubMed]
- Wu, G. Functional amino acids in nutrition and health. Amino Acids 2013, 45, 407–411. [Google Scholar] [CrossRef]
- Wu, G. Functional amino acids in growth, reproduction, and health. Adv. Nutr. 2010, 1, 31–37. [Google Scholar]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar]
- Andersen, S.M.; Waagbo, R.; Espe, M. Functional amino acids in fish nutrition, health and welfare. Front. Biosci. 2016, 8, 143–169. [Google Scholar]
- Gao, Y.; Tian, L.; Liu, Y. Research progress of amino acids in fish. Guangdong Feed. 2012, 21, 53–57. [Google Scholar]
- Wheelhouse, N.M.; Stubbs, A.K.; Lomax, M.A.; MacRae, J.C.; Hazlerigg, D.G. Growth hormone and amino acid supply interact synergistically to control insulin-like growth factor-I production and gene expression in cultured ovine hepatocytes. J. Endocrinol. 1999, 163, 353–361. [Google Scholar] [CrossRef]
- Mihalik, S.J.; Michaliszyn, S.F.; de las Heras, J.; Bacha, F.; Lee, S.; Chace, D.H.; DeJesus, V.R.; Vockley, J.; Arslanian, S.A. Metabolomic profiling of fatty acid and amino acid metabolism in youth with obesity and type 2 diabetes: Evidence for enhanced mitochondrial oxidation. Diabetes Care 2012, 35, 605–611. [Google Scholar] [CrossRef]
- Chalamaiah, M.; Dinesh Kumar, B.; Hemalatha, R.; Jyothirmayi, T. Fish protein hydrolysates: Proximate composition, amino acid composition, antioxidant activities and applications: A review. Food Chem. 2012, 135, 3020–3038. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Al-Tobasei, R.; Lourenco, D.; Leeds, T.; Kenney, B.; Salem, M. Genome-wide association study identifies genomic loci affecting filet firmness and protein content in rainbow trout. Front. Genet. 2019, 10, 386. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Hu, Y.; Zhang, X.; Wei, X.; Wang, T.; Yin, S. Integrated application of multi-omics provides insights into cold stress responses in pufferfish Takifugu fasciatus. BMC Genom. 2019, 20, 563. [Google Scholar] [CrossRef]
- Wei, Z.; Zhou, H.; Zhang, Y.; Zhang, Q.; Zhang, W.; Mai, K. Integrative analysis of transcriptomics and metabolomics profiling on flesh quality of large yellow croaker Larimichthys crocea fed a diet with hydroxyproline supplementation. Br. J. Nutr. 2018, 119, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Mekuchi, M.; Sakata, K.; Yamaguchi, T.; Koiso, M.; Kikuchi, J. Trans-omics approaches used to characterise fish nutritional biorhythms in leopard coral grouper (Plectropomus leopardus). Sci. Rep. 2017, 7, 9372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Fu, W.; Xu, W.; Zhang, H.; Chen, S.; Liu, W.; Peng, L.; Xiao, Y. Comparative transcriptome and DNA methylation analyses of the molecular mechanisms underlying skin color variations in Crucian carp (Carassius carassius L.). BMC Genet. 2017, 18, 95. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Hemani, G.; Vinkhuyzen, A.A.; Chen, G.B.; Lee, S.H.; Wray, N.R.; Goddard, M.E.; Yang, J. Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet. 2014, 10, e1004269. [Google Scholar] [CrossRef]
- Zheng, G.; Jin, W.; Fan, P.; Feng, X.; Bai, Y.; Tao, T.; Yu, L. A novel method for detecting amino acids derivatized with phenyl isothiocyanate by high-performance liquid chromatography-electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 2015, 392, 1–6. [Google Scholar] [CrossRef]
- Statsoft Inc. Statistica Six Sigma, Release 7; Copyright 1984–2006; Statsoft Inc.: Tulsa, OK, USA, 2004. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Malinsky, M.; Svardal, H.; Tyers, A.M.; Miska, E.A.; Genner, M.J.; Turner, G.F.; Durbin, R. Whole-genome sequences of Malawi cichlids reveal multiple radiations interconnected by gene flow. Nat. Ecol. Evol. 2018, 2, 1940–1955. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, W.; Luo, J.; Zhang, D.; Zuhong, L. In Situ bisulfite modification of membrane-immobilized DNA for multiple methylation analysis. Anal. Biochem. 2006, 359, 183–188. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Wang, Z.; Li, X.; Jiang, Y.; Shao, Q.; Liu, Q.; Chen, B.; Huang, D. swDMR: A sliding window approach to identify differentially methylated regions based on whole genome bisulfite sequencing. PLoS ONE 2015, 10, e0132866. [Google Scholar]
- Xu, P.; Zhang, X.; Wang, X.; Li, J.; Liu, G.; Kuang, Y.; Xu, J.; Zheng, X.; Ren, L.; Wang, G.; et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014, 46, 1212–1219. [Google Scholar] [CrossRef]
- Xu, P.; Xu, J.; Liu, G.; Chen, L.; Zhou, Z.; Peng, W.; Jiang, Y.; Zhao, Z.; Jia, Z.; Sun, Y.; et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019, 10, 4625. [Google Scholar] [PubMed]
- Haque, M.M.; Nilsson, E.E.; Holder, L.B.; Skinner, M.K. Genomic clustering of differential DNA methylated regions (epimutations) associated with the epigenetic transgenerational inheritance of disease and phenotypic variation. BMC Genom. 2016, 17, 418. [Google Scholar]
- Zhou, Y.; Luczkowska, K.; Maechler, P. In Situ assessment of hepatic glutamate dehydrogenase activity provides novel insights into amino acid metabolism. FASEB J. 2021, 35, S1. [Google Scholar]
- Dawson, N.J.; Storey, K.B. An enzymatic bridge between carbohydrate and amino acid metabolism: Regulation of glutamate dehydrogenase by reversible phosphorylation in a severe hypoxia-tolerant crayfish. J. Comp. Physiol. B 2012, 182, 331–340. [Google Scholar]
- Seguin, A.; Takahashi-Makise, N.; Yien, Y.Y.; Huston, N.C.; Whitman, J.C.; Musso, G.; Wallace, J.A.; Bradley, T.; Bergonia, H.A.; Kafina, M.D.; et al. Reductions in the mitochondrial ABC transporter Abcb10 affect the transcriptional profile of heme biosynthesis genes. J. Biol. Chem. 2017, 292, 16284–16299. [Google Scholar]
- Gibson, C.; de Ruijter-Villani, M.; Rietveld, J.; Stout, T.A.E. Amino acid transporter expression in the endometrium and conceptus membranes during early equine pregnancy. Reprod. Fertil. Dev. 2018, 30, 1675–1688. [Google Scholar]
- Roberts, M.D.; Romero, M.A.; Mobley, C.B.; Mumford, P.W.; Roberson, P.A.; Haun, C.T.; Vann, C.G.; Osburn, S.C.; Holmes, H.H.; Greer, R.A.; et al. Skeletal muscle mitochondrial volume and myozenin-1 protein differences exist between high versus low anabolic responders to resistance training. PeerJ 2018, 6, e5338. [Google Scholar]
- Ren, R.M.; Liu, H.; Zhao, S.H.; Cao, J.H. Targeting of miR-432 to myozenin1 to regulate myoblast proliferation and differentiation. Genet. Mol. Res. 2016, 15, gmr15049313. [Google Scholar] [CrossRef]
- Adeva-Andany, M.M.; Lopez-Maside, L.; Donapetry-Garcia, C.; Fernandez-Fernandez, C.; Sixto-Leal, C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids 2017, 49, 1005–1028. [Google Scholar]
- Qin, X.; Akter, F.; Qin, L.; Xie, Q.; Liao, X.; Liu, R.; Wu, X.; Cheng, N.; Shao, L.; Xiong, X.; et al. MicroRNA-26b/PTEN signaling pathway mediates glycine-induced neuroprotection in SAH injury. Neurochem. Res. 2019, 44, 2658–2669. [Google Scholar]
- Minich, D.M.; Brown, B.I. A review of dietary (phyto) nutrients for glutathione support. Nutrients 2019, 11, 2073. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (min) | Eluent A (%) | Eluent B (%) |
|---|---|---|
| 0 | 100 | 0 |
| 14 | 85 | 15 |
| 29 | 66 | 34 |
| 30 | 0 | 100 |
| 37 | 0 | 100 |
| 37.1 | 100 | 0 |
| 45 | 100 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zhu, Y.; Xu, P.; Zhao, Z.; Feng, J.; Wu, B.; Jiang, Y.; Dong, C.; Xu, J. Multi-Omics Data Reveal Amino Acids Related Genes in the Common Carp Cyprinus carpio. Fishes 2022, 7, 225. https://doi.org/10.3390/fishes7050225
Zhang H, Zhu Y, Xu P, Zhao Z, Feng J, Wu B, Jiang Y, Dong C, Xu J. Multi-Omics Data Reveal Amino Acids Related Genes in the Common Carp Cyprinus carpio. Fishes. 2022; 7(5):225. https://doi.org/10.3390/fishes7050225
Chicago/Turabian StyleZhang, Hanyuan, Youxiu Zhu, Peng Xu, Zixia Zhao, Jianxin Feng, Biyin Wu, Yanliang Jiang, Chuanju Dong, and Jian Xu. 2022. "Multi-Omics Data Reveal Amino Acids Related Genes in the Common Carp Cyprinus carpio" Fishes 7, no. 5: 225. https://doi.org/10.3390/fishes7050225