
A Detailed Protocol for Constructing a Human Single-Chain Variable Fragment (scFv) Library and Downstream Screening via Phage Display

Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
- SuperScriptTM III First-Strand Synthesis System (Invitrogen, Waltham, MA, USA; Cat. no.: 18080051).
- Oligo(dT)12–18 Primer (Invitrogen, Waltham, MA, USA; Cat. no.: 18418012).
- PlatinumTM Hot Start PCR Master Mix (2×) (Invitrogen, Waltham, MA, USA; Cat. no.: 13000012).
- UltraPureTM DNase/RNase-Free Distilled Water (Invitrogen, Waltham, MA, USA; Cat. no.: 10977015).
- Gel loading dye, purple, 6× (New England Biolabs, Ipswich, MA, USA; Cat. no.: B7024S).
- SeaKem LE agarose (Lonza, Basel, Switzerland; Cat. no.: 50004).
- Ethyl alcohol, pure (Sigma-Aldrich, St. Louis, MO, USA; Cat. no.: E7023).
- Sodium acetate, 3 M, pH 5.2, molecular biology grade (Millipore Sigma, Burlington, MA, USA; Cat. no.: 567422).
- Zymoclean Gel DNA Recovery kit (capped columns) (Zymo Research, Irvine, CA, USA; Cat. no.: D4007/D4008).
- pComb3XSS plasmid vector (Addgene, Watertown, MA, USA; Cat. no.: 63890).
- SfiI restriction enzyme (New England Biolabs, Ipswich, MA, USA; Cat. no.: R0123S).
- T4 DNA ligase (New England Biolabs, Ipswich, MA, USA; Cat. no.: M0202S).
- T4 DNA ligase reaction buffer (New England Biolabs, Ipswich, MA, USA; Cat. no.: B0202S).
- XL1-Blue Electroporation-competent cells (pUC18 plasmid included) (Agilent, Santa Clara, CA, USA; Cat. no.: 200228).
- GlycoBlue Co-precipitant (15 mg/mL) (Invitrogen, Waltham, MA, USA; Cat. no.: AM9516).
- Electroporation cuvettes, 0.2 cm (Invitrogen, Waltham, MA, USA; Cat. no.: P45050).
- 15 mL Falcon conical centrifuge tube (Fisher scientific, Hampton, NH, USA; Cat. no.: 14-959-53A).
- 50 mL Falcon high-clarity conical centrifuge tube (Fisher scientific, Hampton, NH, USA; Cat. no.: 14-432-22).
- SOC medium (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 15544034).
- GeneJET Plasmid miniprep kit (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: K0502).
- Falcon 14 mL round-bottom test tubes with cap (Fisher scientific, Hampton, NH, USA; Cat. no.: 14-959-11B).
- CM13 interference-resistant helper phage (Antibody design labs, San Diego, CA, USA; Cat. no.: PH020L).
- Nunc MaxiSorp ELISA plate (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 442404).
- Immulon 4HBX ELISA plate (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 3855).
- Glucose (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 15023021).
- Ampicillin sodium salt (Millipore Sigma, Burlington, MA, USA; Cat. no.: A9518).
- Carbenicillin disodium salt (Millipore Sigma, Burlington, MA, USA; Cat. no.: C3416).
- Tetracycline (Thermo Fisher scientific, Waltham, MA, USA; Cat. No.: J61714-14).
- Kanamycin disulfate salt (Millipore Sigma, Burlington, MA, USA; Cat. no.: K1876).
- LB broth base powder (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 12780052).
- LB agar powder (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 2270025).
- Sulfuric acid (H2SO4) (Millipore Sigma, Burlington, MA, USA; Cat. no.: 339741).
- Hydrochloric acid (HCl) (Millipore Sigma, Burlington, MA, USA; Cat. no.: 320331).
- Sodium chloride (NaCl) (Millipore Sigma, Burlington, MA, USA; Cat. no. 13423).
- Yeast extract (Millipore Sigma, Burlington, MA, USA; Cat. no.: Y1625).
- Bacto-tryptone (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 211705).
- A 0.2 μm vacuum filter system (Millipore Sigma, Burlington, MA, USA; Cat. no.: Z358193).
- Polyethylene glycol (PEG)-6000 (Millipore Sigma, Burlington, MA, USA; Cat. no.: 807491).
- Tween-20 (Millipore Sigma, Burlington, MA, USA; Cat. no.: P1379).
- Phosphate-buffered saline (PBS) (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: 10010023).
- AEBSF, hydrochloride (Millipore Sigma, Burlington, MA, USA; Cat. no.: 101500)
- Trypsin (Millipore Sigma, Burlington, MA, USA; Cat. no.: T4799).
- M13 phage coat protein monoclonal antibody (clone A5B3), HRP (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: MA5-36125).
- OptEIATM TMB substrate reagent set (BD Biosciences, Franklin Lakes, NJ, USA, Cat. no.: 555214).
- Polycarbonate Erlenmeyer Flask (battled base), 250 mL (Thomas scientific, Chadds Ford Township, PA, USA; Cat. no.: FBC0250S).
- TG1 phage competent cells (Antibody design labs, San Diego, CA, USA; Cat. no.: PC001).
- Falcon 96-well, non-treated round-bottom plate (Fisher scientific, Hampton, NH, USA; Cat. no.: 08-772-54).
- PBMCs isolated from patients’ blood were stored in RNAlater solution at −20 °C.
2.2. Equipment
- C1000 Touch Thermal Cycler with Dual 48/48 Fast Reaction Module (Bio-Rad, Hercules, CA, USA; Cat. no.: 1851148).
- Gene Pulser II Electroporation system (Bio-Rad, Hercules, CA, USA; Cat. no.: 165–2110).
- Vac-Man Laboratory vacuum manifold (Promega, Madison, WI, USA; Cat. no.: A7231).
- FilterMax F5 Multi-Mode Microplate reader (Molecular devices, San Jose, CA, USA; Cat. no.: F5).
- Heidolph Titramax 101 Vibrating platform shaker (Marshall scientific, Hampton, NH, USA; Cat. no.: H036130080).
- Thermo MaxQ 6000 Incubated/Refrigerated shaker (Thermo Fisher scientific, Waltham, MA, USA, Cat. no.: SHKE6000-8CE).
- Thermo MaxQ 8000 Incubated/Refrigerated shaker (Thermo Fisher scientific, Waltham, MA, USA; Cat. no.: SHKE8000-8CE).
3. Procedure
3.1. First Strand cDNA Synthesis (Reverse Transcription, 1 Day)
- Quantify the RNA concentration in the purified RNA. If the RNA concentration is low, the concentration of the RNA is recommended prior to the next steps. Aim for a final concentration of 40 μg of total RNA in 128 μL of ultrapure water. If the concentration exceeds this, dilute the RNA accordingly.
- To the RNA, add 16 μL of 50 μM oligo(dT)12–18 and 16 μL of 10 mM dNTP mix to achieve a final volume of 160 μL. Incubate at 65 °C for 5 min, then place on ice for at least 1 min. Briefly centrifuge to collect any liquid from the cap or side of the tube.
- In a separate RNase-free microcentrifuge tube, prepare a reverse transcription mix by combining 32 μL of 10× RT buffer, 64 μL of 25 mM MgCl2, 32 μL of 100 mM DTT, 16 μL of 40 U/μL RNaseOUT, and 16 μL of 200 U/μL SuperScript III reverse transcriptase for a total of 160 μL.
- Combine the reverse transcription mix with the RNA/Oligo/dNTP mixture to reach a total volume of 320 μL.
- Incubate the mixture at 50 °C for 50 min, then at 85 °C for 5 min. Cool on ice for at least 1 min.
- Add 16 μL of 2 U/μL E. coli RNase H and incubate for 20 min at 37 °C. Centrifuge briefly and pool the resultant first-strand cDNAs from different tubes.
 PAUSE STEP The first-strand cDNA may be stored at −20 °C for several weeks. For
extended storage (several months to years), add 0.1 volume (33.6 μL) of 3 M
sodium acetate (pH 5.2) and 2.2 volume (739.2 μL) of EtOH, vortex to mix well,
and store at −80 °C.
PAUSE STEP The first-strand cDNA may be stored at −20 °C for several weeks. For
extended storage (several months to years), add 0.1 volume (33.6 μL) of 3 M
sodium acetate (pH 5.2) and 2.2 volume (739.2 μL) of EtOH, vortex to mix well,
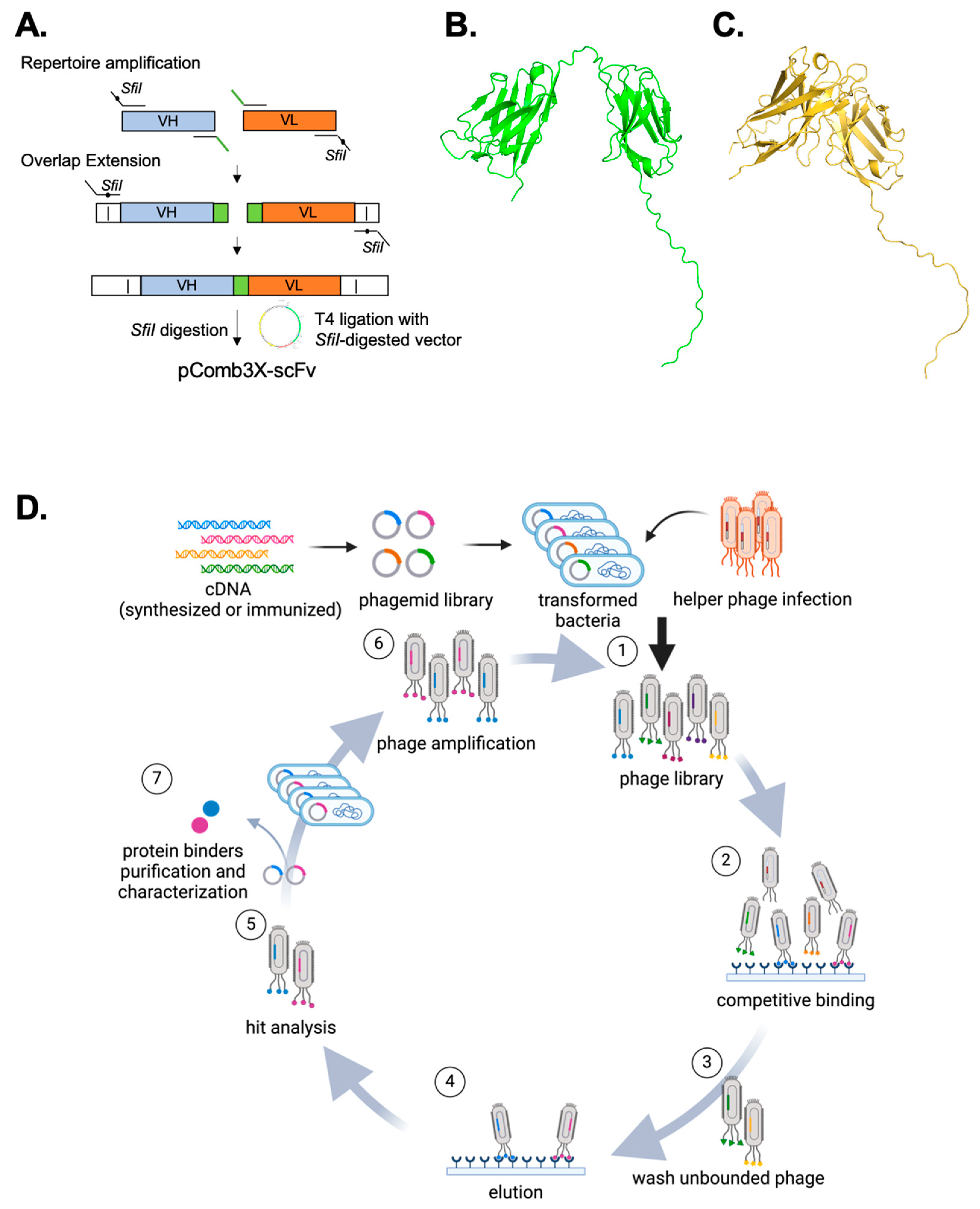
and store at −80 °C.3.2. scFv Construction (6–10 Days)
3.2.1. V Gene Amplification (3–7 Days)
- Prepare 348 unique PCR master mixes, with each PCR primer combination, and conduct Touch Down PCR as follows (Table 2):
- 2.
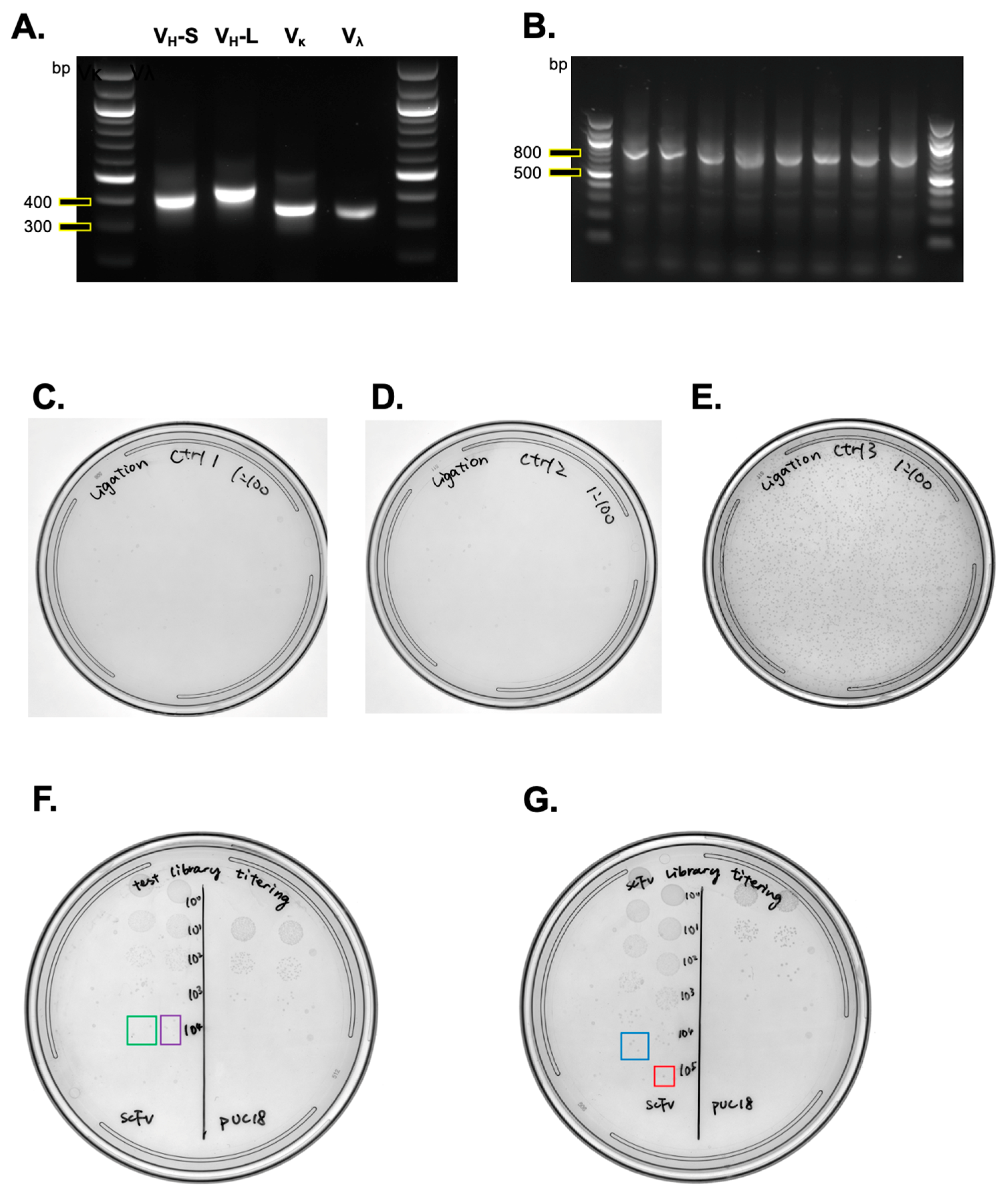
- Verify the PCR amplification success on a 1.5% agarose gel. Combine 3 μL of the PCR product with 2 μL of 6× DNA gel loading dye and perform gel electrophoresis at 135 V for 20 min.
- 3.
- Proceed with ethanol precipitation for PCR products that show successful amplification.
PAUSE STEP The PCR product can be stored at 4 °C for up to 7 days if
not proceeding immediately.3.2.2. Ethanol (EtOH) Precipitation of Pooled PCR Reactions (1 Day)
- Combine the PCR products for each V region into a separate 15 mL Falcon tube, ensuring you prepare four tubes for the four V regions. Mix thoroughly, then aliquot 330 μL into individual 1.5 mL microcentrifuge tubes.
 CRITICAL STEP It is essential to ensure a consistent mix
in the 15 mL Falcon tubes before aliquoting, to maintain diversity and
homogeneity, as some DNA might be lost during the EtOH precipitation process.
CRITICAL STEP It is essential to ensure a consistent mix
in the 15 mL Falcon tubes before aliquoting, to maintain diversity and
homogeneity, as some DNA might be lost during the EtOH precipitation process.- 2.
- Add 0.1 volume (33 μL) of 3 M sodium acetate to each tube, followed by 3 volumes (1089 μL) of 100% EtOH. Incubate at −20 °C overnight to precipitate the DNA.
- 3.
- Centrifuge at 16,000× g for 10 min at room temperature (RT) and aspirate most of the EtOH without disturbing the pellet.
- 4.
- Wash the DNA pellet with 1 mL of ice-cold 70% EtOH (in ultrapure water) and centrifuge at 16,000× g for 2 min at RT. Carefully aspirate the ethanol without disturbing the pellet. Repeat this wash.
CRITICAL STEP Ice-cold 70% EtOH is recommended to
prevent DNA loss, as DNA may dissolve in the aqueous portion of RT 70% EtOH.- 5.
- After the second wash, centrifuge at 16,000× g for 2 min at RT and remove all the EtOH.
- 6.
- Air-dry the pellet for 15 min to an hour with the tube lid open to ensure it is completely dry.
- 7.
- Resuspend the DNA pellet from each tube in 30 μL ultrapure water (preheated to 55 °C), and pool the resuspensions into four 4 tubes (one for each V fragment).
- 8.
- Run 25 μL of each resuspended V fragment DNA on a 1.5% agarose gel at 100 V for 40 min, using multiple lanes as needed to fit all samples.
CRITICAL STEP Employ low voltage for gel electrophoresis
to prevent heat-induced DNA damage.- 9.
- Extract DNA from the gel using a Zymoclean Gel DNA Recovery kit and elute in water heated to 55 °C to maximize yield. Determine DNA using a spectrophotometer.
CRITICAL STEP This
step is crucial to diminish primer interference from the first PCR in
subsequent amplifications. PAUSE STEP Store the
purified DNA at −20 °C for long-term storage.3.2.3. Secondary Overlapping Extension (SOE) PCR (2 Days)
- Use 50 ng of each VH and VL, along with 5 μL of primer mix, for the reactions. We recommend performing 12 SOE reactions for each construct to ensure an adequate quantity of DNA is produced. Execute the PCR according to the protocol outlined in Table 3.
CRITICAL STEP Performing 20 cycles prior to an infinite
hold at 12 °C is crucial. Fewer than 20 cycles may result in indistinct scFv
bands, while more than 20 cycles can impair polymerase activity.- 2.
- Assess the SOE PCR success by electrophoresing 3 μL from each of the 48 tubes on a 1.5% agarose gel.
- 3.
- Pool the remaining SOE PCR products into four separate microcentrifuge tubes.
- 4.
- Repeat DNA EtOH precipitation as described in Section 3.2.2, “EtOH precipitation of pooled PCR reactions.” Combine approximately 360 μL of the reaction mixture from 12 PCR reactions per construct, adding 0.1 volume (30 μL) of 3 M sodium acetate, followed by 3 volumes (1080 μL) of 100% EtOH.
- 5.
- Precipitate DNA overnight at −20 °C.
- 6.
- The following day, wash the precipitated DNA as outlined in Section 3.2.2, “EtOH precipitation of pooled PCR reactions.”
- 7.
- Utilize the required amount for digestion as detailed in Section 3.3, “Cloning scFv constructs to pComb3XSS by SfiI digestion and T4 DNA ligase reaction,” or store the remainder of the DNA.
PAUSE STEP The precipitated DNA can be stored at −20 °C for several
weeks.3.3. SfiI Digestion of pComb3XSS and scFv Constructs (2 Days)
- Digest 50 μg of the pComb3XSS vector with 10 μL of SfiI enzyme (4 units/μg of DNA), bringing the total volume to 250 μL with water (50 μL for every 10 μg of DNA).
- Digest 15 μg of the scFv construct precipitated from the prior step with 3 μL of SfiI enzyme, adjusting the final volume to 150 μL with water (50 μL for every 5 μg of DNA).
- Incubate the digestion reactions at 37 °C overnight.
- OPTIONAL Despite the manufacturer’s recommendations for a 60 °C reaction temperature, 37 °C may be used for stable and prolonged digestion.
- Confirm complete digestion by comparing the digested fragments with the undigested vector on a 1.5% agarose gel.
- Use a Zymoclean Gel DNA Recovery kit to gel-extract the fully digested pComb3Xss (3379 bp), the stuffer fragment (1673 bp—removed from the pComb3XSS), and scFv constructs (700–800 bp). Elute with water preheated to 55 °C to maximize yield.
PAUSE STEP Store the eluted DNA at −20 °C for several weeks.3.4. Test Library Generation (2 Days)
3.4.1. Single T4 DNA Ligase Reaction of scFv into pComb3xSS (Adapted from [14], 1 Day)
- Prepare four ligation reactions as follows, which include three controls and the scFv ligation reactions for each construct:
- Control #1: without ligase;
- Control #2: without insert;
- Control #3: with stuffer insert;
- Four separate scFv ligations for each scFv construct
- The composition and volumes for these reactions are detailed in the table provided (Table 4).
- Carry out the T4 DNA ligase reaction according to the protocol specified in the subsequent table (Table 4).
- 3.
- On the following day, combine 1 μL from each of the four scFv construct ligation reactions (totaling 20 μL) into a new microcentrifuge tube, resulting in a total volume of 4 μL. This mixture will be electroporated into 50 μL of XL1-Blue competent cells.
- 4.
- OPTIONAL STEP For quality control of individual scFv constructs, heat-shock TOP10 competent cells with 1 μL of each ligation product (four scFvs separately) and perform a dilution series inoculation on LB agar plates supplemented with ampicillin to assess diversity size.
3.4.2. Test Library Electroporation (1 Day)
- In a new 1.5 mL microcentrifuge tube, combine 1 μL from each of the four scFv ligation products for electroporation into 50 μL of XL1-Blue competent cells.
- Pre-warm SOC media to 37 °C and prepare 2 mL of pre-warmed SOC media in 15 mL Falcon tubes.
- Thaw 250 μL of XL1-Blue competent cells, allocating 50 μL for each of the five electroporation samples. These include three negative controls (no ligase, no insert control, and stuffer insert) and the combined scFv sample (4 μL total), along with a pUC18 electroporation control.
- Aliquot 50 μL of competent cells into each pre-cooled microcentrifuge tube: mix 50 μL of competent cells with 4 μL of the ligation product for each control and scFv, along with 1 μL of 0.1 ng/μL pUC18 plasmid as a positive control. Mix gently by pipetting and transfer into 0.2 cm gap electroporation cuvettes, avoiding bubble formation.
- Set the Gene Pulser II apparatus to 25 μF capacitance, 2.5 kV, and 200 Ohm.
- Tap the cuvette gently to remove bubbles and ensure all cells settle at the bottom, free from bubbles.
CRITICAL STEP The pulse should result in 12.5 kV/cm with a time constant
of 4–5 milliseconds.- 7.
- Immediately add 950 μL of pre-warmed SOC to the electroporated cuvette, and transfer the mixture to the prepared 15 mL Falcon tubes containing 2 mL of SOC, making a total volume of 3 mL. Incubate at 37 °C with shaking at 225 rpm for 1 h. Meanwhile, pre-warm four LB agar plates supplemented with ampicillin to 37 °C.
- 8.
- After 1 h of recovery, take 10 μL from the transformed cells and from the pUC18-transformed cells to estimate the library size. Perform tenfold serial dilutions up to 10−8 in a 96-well plate. Drop 5 μL from each dilution onto a pre-warmed LB agar plate with ampicillin using a multichannel pipette or a 96-pin pronger. Plate the three ligation controls (no ligase, no insert, and stuffer insert) at a 1:100 dilution on separate LB agar plates.
- 9.
- Incubate all plates at 37 °C overnight (approximately 16 h).
- 10.
- The following day, calculate the library size from the test library using the formula:
3.5. Direct (Large-Scale) Library Generation (3 Days)
3.5.1. Large-Scale Ligation (1 Day)
- Conduct multiple ligation reactions to reach the target library > 107 size.
- Follow the T4 DNA ligase reaction protocol as outlined in Section 3.4.1, “Single T4 DNA ligase reaction of scFv into pComb3XSS.”
3.5.2. EtOH Precipitation of the Ligated scFv Phagemid Library (1 Day)
- Pool the ligation reactions up to a maximum of 330 μL per tube. Add 0.1 volume of 3 M sodium acetate and 3 volumes of EtOH, as described in Section 3.2.2, “EtOH precipitation of pooled PCR reactions.”
- 2.
- The next day, centrifuge the precipitated DNA and wash the pellet following the steps in Section 3.2.2, “EtOH precipitation of pooled PCR reactions.” Resuspend the pellet in a total of 200 μL of ultrapure water warmed to 55 °C.
3.5.3. Electroporation into XL1-Blue Competent Cells (1 Day)
- Pre-cool the electroporation adaptor, a 15 mL Falcon tube, a microcentrifuge tube, and electroporation cuvettes (0.2 cm) on ice.
- Pre-warm 50 mL Falcon tubes containing 3 mL of SOC medium and 5 mL of SOC medium to 37 °C.
- Prepare the required volume of 50 μL competent cell mixed with 14.6 μL DNA (based on calculations to reach the desired library size), mixing slowly for thorough integration.
- Prepare a control mixture with pUC18 and competent cells in a separate microcentrifuge tube.
- Aliquot the scFv master mix into the electroporation cuvettes, ensuring the absence of air bubbles. Wipe the cuvettes’ metal surfaces to remove moisture.
- Repeat the electroporation process as in Section 3.4.2, “Test library electroporation.”
- Immediately add 935 μL of warmed SOC media to each cuvette and transfer the bacteria to the pre-warmed 50 mL Falcon tubes. Ensure each tube does not exceed 10 mL of total volume for adequate aeration.
CRITICAL STEP The volume of bacteria in the Falcon tube should not
exceed 20% of the tube’s volume to aerate bacteria for growth. Thus, add only
up to 7 electroporated bacteria (each 1 mL) to each 50 mL Falcon tube
containing 3 mL of SOC media.- 8.
- Shake the electroporated bacteria at 37 °C and 225 rpm for 1 h for recovery.
- 9.
- For library size calculation, dilute 100 μL from the Falcon tube for serial dilutions to 10−8, then plate 5 μL from each dilution. Store the remaining dilutions at 4 °C.
- 10.
- Spin down the rest of the bacteria and resuspend in a total of 4 mL. Incubate 1 mL onto each of several 245 mm square LB agar plates (with 2% w/v glucose and 50 μg/mL ampicillin).
- 11.
- Incubate the plates overnight at 37 °C.
- 12.
- Scrape the bacteria from the plates using 4 mL of LB media, rinsing with an additional 2 mL of LB. Be gentle to avoid scraping the agar.
- 13.
- Add glycerol to a final concentration of 20%. Dilute 100 μL of the scraped bacteria in 900 μL LB and measure the OD at 600 nm (OD600). An OD600 corresponds to 1 × 108 bacteria per mL.
CRITICAL STEP Dilute the bacteria before measuring OD600 to
avoid exceeding the spectrophotometer’s detection limit.- 14.
- Aliquot the bacteria to achieve 5 × 109 bacteria per cryovial for storage at −80 °C.
PAUSE STEP The scFv bacteria library can be stored at −80 °C for
years.- 15.
- OPTIONAL STEP Adjust the number of bacteria per vial based on library diversity. A higher number of bacteria based on library diversity can be used so that each library stock contains 48 or more copies per scFv repertoire [35]. For example, if the diversity size is 8 × 108, 4 × 1010 bacteria can be aliquoted to each cryovial so that each stock contains 50 copies of each scFv in the library. However, avoid exceeding 1 × 1012 bacteria per vial to prevent spontaneous phage precipitation.
3.6. Library Quality Assessment (2–3 Days)
- Thaw a vial of the library stock and prepare a serial dilution series to 10−8.
- Inoculate 100 μL of each dilution onto LB agar plates containing 2% w/v glucose, 50 μg/mL ampicillin.
- 3.
- Incubate the plates at 37 °C overnight to allow colony growth.
- 4.
- Pick individual colonies and inoculate them into 3 mL of LB media supplemented with ampicillin, using Falcon 14 mL round-bottom tubes.
- 5.
- Incubate these cultures at 37 °C overnight to promote further colony growth.
- 6.
- Perform a miniprep using the GeneJET Miniprep kit to extract bacterial plasmid.
- 7.
- Sequence the extracted DNA using Sanger sequencing with the sequencing primers provided in Appendix A.
3.7. Phage Display Affinity Selection (Variable Time Depending on Enrichment, Usually 14–20 Days)
3.7.1. Optimization of Antigen Coating Condition on Different ELISA Plates (1–2 Days)
- Select appropriate ELISA plates for optimal coating of the target protein.
- Set various antigen coating concentrations from 1–5 µg/mL based on the manufacturer’s guidelines. Target and control antigen proteins should be in similar forms.
- Coat the ELISA plates, 100 µL per well, with triplicates of both target and control antigens in PBS (or an appropriate buffer for the antigens) and incubate overnight at 4 °C with gentle shaking.
- Wash the plates three times with PBS-T (0.05% Tween-20 in PBS).
- Block the plates using 200 μL of 5% milk in PBS-T, incubating for 1 h at RT with gentle shaking.
- Wash the plates three times with PBS-T.
- Add 100 μL of HRP-conjugated antibody specific to the target or tag of the target protein and control antigens. Follow the manufacturer’s guidelines and incubate for 1 h at RT with gentle shaking.
- 8.
- Wash the plates three times with PBS-T.
- 9.
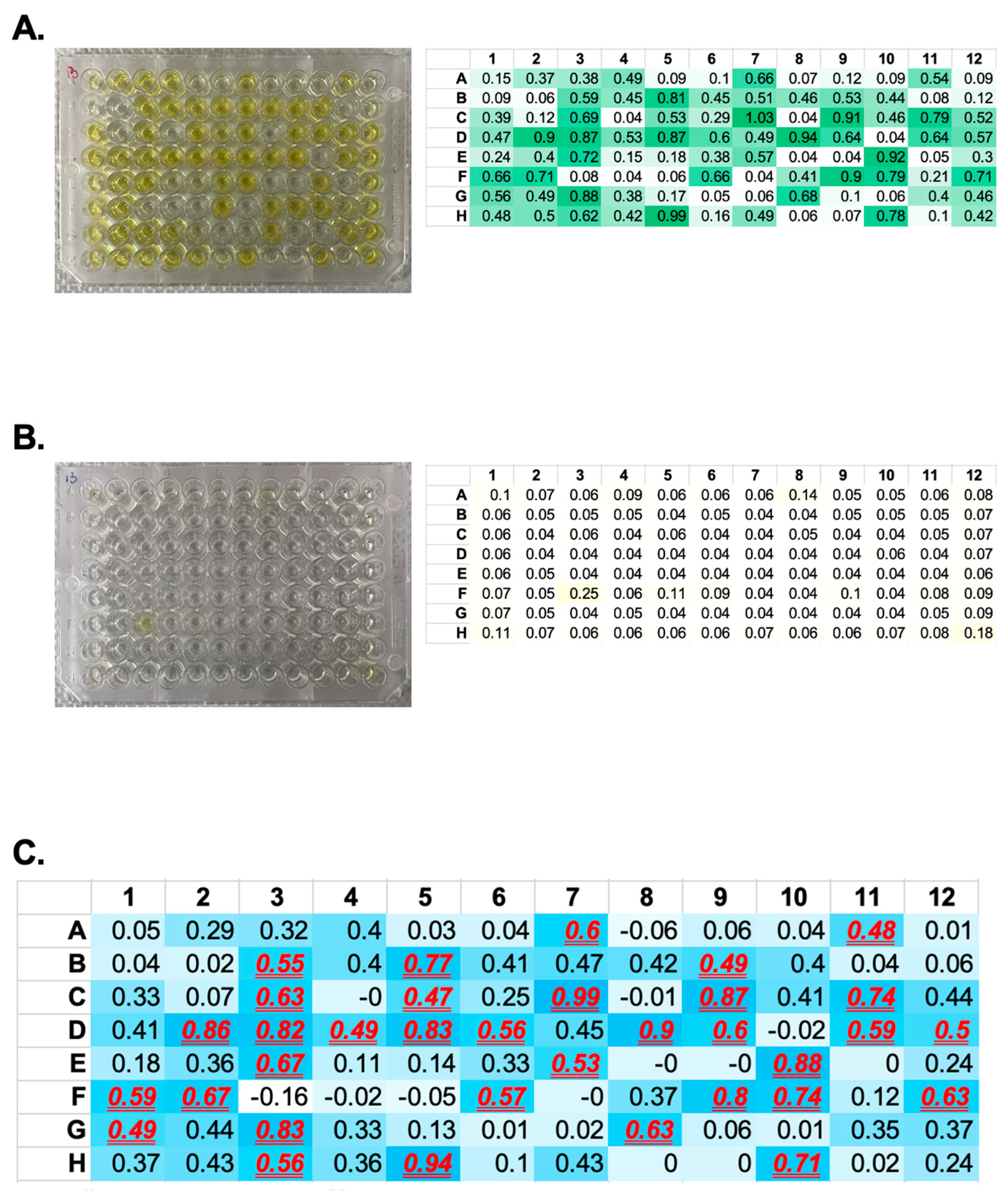
- Develop the reaction with 100 μL of TMB substrate for 5 to 20 min, then stop the reaction with 50 μL of 1 M H2SO4 (or 1 N HCl). Measure the signal intensity at 450 nm.
- 10.
- Plot a titration curve of OD450nm against different antigen coating concentrations to find the optimal concentration for affinity selection.
3.7.2. Induction of Phages Carrying scFv and In Vitro Selection (Multiple Rounds of 3 Days)
- Inoculate the library stock into 60 mL of 2xTY-G supplied with 50 µg/mL ampicillin (and 50 µg/mL tetracycline for XL1-Blue cells). Rinse the tube to collect all bacteria.
- Reamplify the bacteria library culture at 37 °C and 200 rpm until OD600 reaches 0.5.
- Transfer 10 mL of the exponential-phase bacteria to a 50 mL Falcon tube and infect with 4 × 1010 cfu of VCSM13 helper phage. Incubate for 30 min at 37 °C without shaking.
CRITICAL STEP Use filtered pipette tips and disinfect the tips before
disposal to prevent phage contamination. Used pipette tips should be discarded
in a biohazard bag for either dispensing or autoclaving later.- 4.
- Centrifuge the cells at 4000× g for 10 min at RT. Discard the supernatant, and briefly place the tube on a paper towel.
- 5.
- Resuspend in 60 mL of 2xTY-I and transfer to a disposable 250 mL baffled flask. Incubate overnight at 37 °C and 200 rpm to induce the phages carrying scFv.
- 6.
- The next day, start growing naïve TG1 (not carrying the scFv gene) in 10 mL LB media in a 50 mL Falcon tube. Grow until OD600 reaches 0.5 to 0.6, then keep in ice.
CRITICAL STEP TG1
cells should be first cultured in M9 medium to ensure the presence of F-factor.- 7.
- Centrifuge the induced 50 mL culture at 4000× g for 30 min at 4 °C.
- 8.
- Transfer 40 mL of supernatant to a new 50 mL Falcon tube, then add 10 mL of PEG solution. Mix well by inverting the tube 5 times.
- 9.
- Incubate the mixture in ice for an hour to precipitate the phages.
- 10.
- Spin down at 4000× g for 30 min at 4 °C. Discard the supernatant and place the tube upside down on a paper towel briefly to remove residual supernatant.
- 11.
- Resuspend the precipitated phage pellet using 1 mL of ice-cold PBS, then transfer the resuspension to a new ice-cold microcentrifuge tube.
- 12.
- Centrifuge at 20,000× g for 1 min at 4 °C to spin down residual bacteria.
- 13.
- Carefully transfer only the supernatant to a new ice-cold microcentrifuge tube.
- 14.
- Add 250 μL of ice-cold PEG solution and pipet up and down several times to mix well.
- 15.
- Incubate on ice for 10 min.
- 16.
- Spin down the phages at 14,000× g for 15 min at 4 °C. Discard the supernatant and resuspend the pellet using 1 mL of ice-cold PBS.
- 17.
- Centrifuge at 14,000× g for 1 min at 4 °C to completely remove residual bacteria.
- 18.
- Transfer the supernatant to a new ice-cold microcentrifuge tube.
- 19.
- To a round bottom 96-well plate, add 90 μL of PBS to a row of 12 wells. Use this PBS to make 12 tenfold serial dilutions of the precipitated phage, beginning with 10 μL of precipitated phage. Serial dilution can be alternatively performed using a multichannel pipettor if preferred. In a separate row, add 90 μL of naïve TG1 (at OD600 = 0.5~0.6) and 10 μL from each serial dilution to achieve a final volume of 100 μL. Incubate the plate at 37 °C for 30 min without shaking.
- 20.
- Carefully pipette 5 μL drops of the infected TG1 onto LB agar plates containing 2% glucose and ampicillin. As a control, also pipette 5 μL of naïve TG1 (uninfected by phages). Allow the drops to air-dry and incubate the plates at 37 °C overnight. Concurrently, inoculate a naïve TG1 into 5 mL of LB media (without antibiotics) in a 50 mL Falcon tube for overnight growth.
CRITICAL STEP When culturing the naïve TG1 cells, also inoculate TG1
into two tubes containing LB with ampicillin or kanamycin to check for
potential contamination by either the phage, helper phage, or both. TG1
contaminated with the induced phage will grow in ampicillin-supplemented media,
and TG1 contaminated with helper phage will grow in kanamycin-supplemented
media.- 21.
- For antigen coating on ELISA plates, use the previously optimized concentration (as per Section 3.7.1, “Optimization of antigen coating condition on different ELISA plates.” Coat four wells with the control antigen and four non-adjacent wells with the target antigen, using 100 μL for each well. Seal the plate with impermeable film and incubate overnight at 4 °C.
- 22.
- The next day, calculate the colony-forming units (cfu) of the precipitated phage using the following formula:
CRITICAL STEP The cfu/mL should be higher than 1 × 1012 cfu/mL,
which would be at least a hundred times greater than the initial bacterial
count. If it is lower, repeat the phage production process. PAUSE STEP Store the precipitated phages at 4 °C for weeks. For
long-term storage, add glycerol to achieve a final concentration of
approximately 40%. The phage in glycerol can then be stored at −80 °C for
several months to years.- 23.
- Inoculate naïve TG1 into 10 mL of LB media in a 50 mL Falcon tube. Grow at 37 °C and 220 rpm until it reaches OD600 = 0.5. Then, move the Falcon tube into an ice bucket until it is ready for infection with the eluted phages.
- 24.
- Discard the antigen-coating solution from the ELISA plates and wash three times with PBS-T. Block the plates by adding 200 μL of 5% milk in PBS-T to each well, and incubate at RT for 1 h with gentle shaking.
- 25.
- During the 1 h blocking, pre-block the phages by adding 5 × 1011 cfu of the precipitated phages to 500 μL of 5% milk in PBS-T (1 × 1011 per well).
CRITICAL STEP The actual required phage volume is 4 × 1011
cfu in 400 μL, but we recommend preparing 5 × 1011 cfu in 500 μL to
compensate for potential transfer loss. The pre-blocking should be performed
for at least 20 min or until the 1 h ELISA plate blocking is complete, with
gentle shaking at RT.- 26.
- After blocking the ELISA plates, wash them three times with PBS-T. Transfer 100 μL of the pre-blocked phages to the wells coated with control antigen for negative selection.
CRITICAL STEP To prevent the target-antigen-coated wells from drying
out, add 100 μL of the blocking solution to the wells coated with the target
antigen.- 27.
- Agitate the plates gently at 700 rpm on a shaking platform for 2 h.
- 28.
- After incubation, remove the blocking solution from wells that have been coated with the target antigen, then transfer the phages from the wells coated with the control antigen to those coated with the target antigen.
CRITICAL STEP To prevent the controlled-antigen-coated wells from drying
out, add 100 μL of the blocking solution to the wells initially coated with the
control antigen.- 29.
- Agitate again at 700 rpm for 2 h.
- 30.
- After agitation, remove the liquid from all eight wells (four coated with control antigen and four with target antigen). Wash all wells 15 times with 200 μL of PBS-T, followed by a single wash with 200 μL of PBS. Add 50 μL of trypsin (0.25 mg/mL in PBS) to all eight wells and agitate at 700 rpm for 30 min at RT. The addition of trypsin facilitates the dissociation of phages from the target antigen by cleaving the digestion sites between the phage coat protein and the displayed scFv. Phages eluted from both control and target antigens should not be discarded, as they are used in the following steps for enrichment analysis.
CRITICAL STEP Liquid waste containing phage particles
must be treated with a disinfectant before being disposed of at a liquid
handling station equipped for regular disinfection.- 31.
- Prepare two microcentrifuge tubes, each containing 10 μL of AEBSF. Transfer the trypsin-eluted phages from the four wells initially coated with control antigen into the first tube, and those from the target antigen wells into the second tube.
- 32.
- In a new 50 mL Falcon tube, add 3 mL of exponential-phase naïve TG1 (from step 23). Infect this culture with 50 μL of the eluted phage from the wells initially coated with target antigen. Incubate the mixture at 37 °C without shaking for 30 min. Then, add 7 mL of LB supplemented with ampicillin and glucose to achieve a final concentration of 50 μg/mL ampicillin and 2% glucose in 10 mL. Incubate the 10 mL mixture overnight at 37 °C and 180 rpm.
- 33.
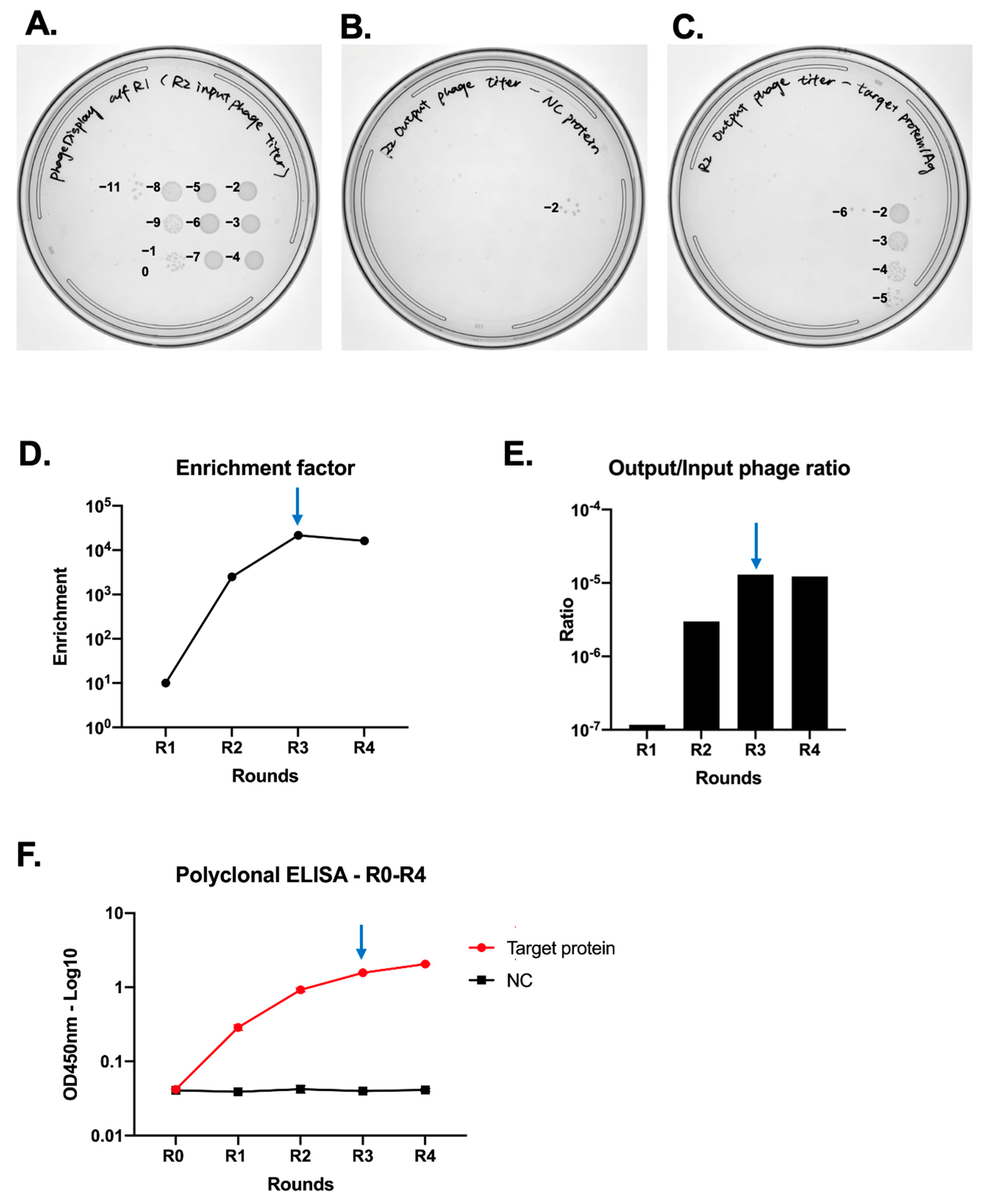
- Perform tenfold serial dilutions of trypsin with AEBSF mixtures from control and target Ag, referring to Section 3.7.3, “Enrichment analysis.” If the enrichment factor is less than 1000 (from the first method) but still increasing (from the second method), proceed to the next round of affinity selection.
- 34.
- The next day, transfer 500 μL of the overnight-grown 10 mL culture into 50 mL of 2xTY-G (2% glucose, ampicillin) in a baffled disposable flask and incubate at 37 °C, 200 rpm.
PAUSE STEP Aliquot the remaining 10 mL of culture into cryovials,
dispensing 1 mL into each vial. Then, add 666 μL of 50% glycerol to each vial
to make the final 20% glycerol, resulting in a total volume of 1.666 mL per
cryovial. These can be stored at −80 °C for future single colony picking or for
restarting the affinity selection process in case of error.3.7.3. Enrichment Analysis (1 Day)
- Prewarm the LB agar plate (2% w/v glucose and 50 μg/mL ampicillin) to 37 °C.
- In a 96-well plate, add 90 μL of PBS to two nonadjacent rows of 12 wells each, making 24 wells in total. Use this to prepare 12 tenfold serial dilutions, starting with 10 μL of the trypsin + AEBSF mixture for both control and target antigens.
- In two separate rows of 12 wells each, add 90 μL of exponential-phase naïve TG1 (from Section 3.7.2, “Induction of phages carrying scFv and in vitro selection,” step 23).
- Add 10 μL of each serial dilution to the 90 μL of naïve TG1.
- Incubate the plate at 37 °C for 30 min without shaking.
- Using a multichannel pipet, inoculate 5 μL from each well of phage-infected TG1 onto multiple LB agar plates (2% glucose and ampicillin). Use three plates each for the control and target antigens to calculate the average values, considering the sticky nature of phages. Include a control spot of 5 μL of naïve TG1 not infected with phages as a sentinel against contamination by phage.
- Incubate the plates overnight at 37 °C.
- The next day, count the average number of colonies from control and target antigens. Calculate the enrichment factor as follows:
- 9.
- An enrichment factor exceeding 1000 is indicative of successful enrichment.
- Determine cfu from the LB agar plates with drops of TG1 infected with phages eluted from wells coated with the target antigen. For example, if there are three average colonies at the 2nd dilution point (this is 10−3 dilution, making the colony number 3 × 103), cfu is
- Given that 4 × 1011 cfu in 400 μL was used for input (equivalent to 1 × 1012/mL), calculate the ratio as .
- This ratio should steadily increase, then plateau or decrease.
CRITICAL STEP The optimal round to conclude affinity selection and
proceed to monoclonal ELISA is the last round before the plateau or decrease in
both methods.3.7.4. Polyclonal ELISA (2 Days)
- Coat the ELISA plate with control and target antigens at the previously optimized concentration, using 100 μL per well for triplicates. Adjust based on the number of time points being tested in the polyclonal ELISA.
- Incubate the plate overnight at 4 °C.
- Wash the plate three times with PBS-T, then block using 200 μL of 5% milk in PBS-T per well for 1 h at RT with gentle shaking.
- During the blocking period, add 1 × 1011 cfu of phages (based on cfu calculation from Enrichment Analysis Method 2) before affinity selection and after each selection round to 5% milk in PBS-T, preparing 100 μL for each time point.
- After blocking, wash the plates three times with PBS-T and transfer 100 μL of phage in milk to each well.
- Incubate for 1 h at RT with gentle shaking.
- Wash the plate three times with PBS-T, add 100 μL of HRP-conjugated anti-M13 coat protein antibody according to the manufacturer’s guidelines, and incubate for 1 h at RT with gentle shaking.
- Wash the plate three times with PBS-T, then add 100 μL of TMB substrate.
- Develop the color reaction, then add 50 μL of 1 M H2SO4 to stop it. Quantify the optical density at 450 nm.
- Draw a titration curve for OD450. If the OD450 value plateaus or decreases, affinity selection should be halted at that round. The bacterial stock from the round immediately preceding the plateau or decrease can be used for single colony picking.
3.8. Screening for Hits by Monoclonal ELISA (4 Days)
- Review the results of enrichment analyses (output/input calculation, enrichment factor analysis, and polyclonal ELISA). A plateau or decrease in output/input calculations and polyclonal ELISA, along with an enrichment factor over 1000, indicate it is time to stop affinity selection. For instance, if these criteria are met by the product of Round 3, proceed to pick single colonies from Round 3.
- Thaw a vial from the −80 °C bacteria stock and prepare a serial dilution series from 10−1 to 10−8. Inoculate each dilution onto LB agar plates (containing 2% glucose and 50 μg/mL ampicillin) to determine the optimal dilution for picking single colonies. Avoid too many or too few colonies on the plates. Incubate the plates overnight at 37 °C.
- The next day, visually assess the plates to determine which dilution yields a manageable number of colonies for picking.
- Plate multiple (10 to 15) LB agar plates (2% glucose and 50 μg/mL ampicillin). Ensure even spreading and drying of the plates to prevent clumping of colonies.
- Prepare 6 to 12 round-bottom 96-well plates, adding 200 μL of 2xTY-G (2% glucose, 50 μg/mL ampicillin) to each well.
- Using a sterile P200 pipette tip, pick single colonies and inoculate them into the prepared wells.
- 7.
- Secure the round-bottom plates in a plastic box affixed to a shaking platform inside an incubator. Remove the covers from the plates, seal the box with cling film, and incubate the bacteria overnight at 37 °C, 250 rpm.
CRITICAL STEP To prevent cross-contamination, keep the plates covered
while securing them inside the box, and only remove the 96-well plate covers
just before sealing the box with cling film.- 8.
- The following day, prepare a new set of 96-well round-bottom plates with 200 μL of 2xTY-G. Transfer 5 μL from the overnight cultures into the new plates using a multichannel pipette.
PAUSE STEP The original 96-well plate with overnight-grown colonies
is the ‘master plate.’ Remove 45 μL from the master plate to prevent expansion of
the media during freezing, add 100 μL of 50% glycerol to achieve a 20% final
glycerol concentration, and store at −80 °C for future use.- 9.
- After 3 h of growth, add 50 μL of 2xTY-G containing 4 × 108 cfu of helper phages to each well, gently mixing to ensure even distribution of the phages with the bacteria.
- 10.
- Incubate at 37 °C for 1 h without shaking to allow for helper phage infection. Then, centrifuge the plates at 4000× g for 10 min and discard the supernatant carefully to prevent cross-contamination between wells, by a single vigorous downward motion. Blot the plates on a paper towel and resuspend the bacterial pellets in 200 μL of 2xTY-I, ensuring thorough mixing.
- 11.
- Return the plates to the incubator and induce phage production overnight at 25 °C and 250 rpm.
- 12.
- Prior to leaving for the day, coat additional ELISA plates with control and target antigens using the previously optimized conditions. Prepare an equal number of plates for each set of monoclonal phages picked.
- 13.
- The next day, wash the ELISA plates three times with PBS-T and block them with 200 μL of 5% milk in PBS-T with gentle shaking for 1 h.
- 14.
- During the blocking step, centrifuge the phage induction plates at 4000× g for 10 min. Transfer 60 μL of the supernatant to new round-bottom 96-well plates containing 180 μL of 5% milk in PBS-T.
- 15.
- After blocking, add 100 μL of the diluted phage solution to each corresponding well in the ELISA plates. Incubate for 1 h (or 2 h to increase the ELISA signal) at RT with gentle agitation.
- 16.
- Wash the plates three times with PBS-T, then add 100 μL of HRP-conjugated anti-M13 coat protein antibody, following the manufacturer’s dilution guidelines. Incubate for 1 h at RT with gentle shaking.
- 17.
- Wash the plate five times with PBS-T and add 100 μL of TMB substrate to develop the color. Stop the reaction with 50 μL of 1 M H2SO4 and quantify the signal of OD450 using a plate reader.
- 18.
- Normalize the ELISA results by subtracting the control antigen quantifications from those of the target antigen. Select colonies with normalized values exceeding a predetermined cutoff for further assays specific to the project.
CRITICAL STEP High ELISA signals, even after normalization, do not
guarantee strong binding in other assays, such as FACS of Western blot using
phages carrying scFv. Therefore, it is recommended to use a low cutoff value,
such as ‘top 33%’, for leniency in determining which colonies to further
analyze.- 19.
- Concurrently with hit selection, perform sequencing of the selected hits. This can be done by inoculating bacteria from the frozen master plate into 3 to 5 mL of LB media supplemented with ampicillin in a 14 mL tube for miniprep or colony PCR. Standard Sanger sequencing can then be carried out to identify the sequence of the hits, with primer details provided in Appendix A. Identify unique hits to proceed with further specific assays.
4. Expected Results
4.1. scFv Library Generation
4.2. Phage Display Affinity Selection and Antigen-Specific Antibody Development
5. Reagents Setup
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Primers for scFv Library Generation
- VH F′ primers (nineteen in total)
- VH R′ short linker primers (VH-S R′, six in total)
- VH R′ long linker (VH-L R′, six in total)
- Vκ (kappa light chain) F′ (twelve in total)
- Vκ (kappa light chain) R′ (five in total)
- Vλ (lambda light chain) F′ (twenty in total)
- Vλ (lambda light chain) R′ (three in total)
- SOE—Secondary Overlapping Extension (two):
- Recommended sequencing primer (for pComb3XSS phagemid):
References
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J.T. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Nahta, R.; Esteva, F.J. Trastuzumab: Triumphs and tribulations. Oncogene 2007, 26, 3637–3643. [Google Scholar] [CrossRef]
- Zhao, S.; Chadwick, L.; Mysler, E.; Moots, R.J. Review of Biosimilar Trials and Data on Adalimumab in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2018, 20, 57. [Google Scholar] [CrossRef]
- Montgomery, H.; Hobbs, F.D.R.; Padilla, F.; Arbetter, D.; Templeton, A.; Seegobin, S.; Kim, K.; Campos, J.A.S.; Arends, R.H.; Brodek, B.H.; et al. Efficacy and safety of intramuscular administration of tixagevimab–cilgavimab for early outpatient treatment of COVID-19 (TACKLE): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Respir. Med. 2022, 10, 985–996. [Google Scholar] [CrossRef]
- Wang, S.S.; Yan, Y.S.; Ho, K. US FDA-approved therapeutic antibodies with high-concentration formulation: Summaries and perspectives. Antib. Ther. 2021, 4, 262–272. [Google Scholar] [CrossRef]
- Stone, C.A., Jr.; Spiller, B.W.; Smith, S.A. Engineering Therapeutic Monoclonal Antibodies. J Allergy Clin Immunol 2023. [Google Scholar] [CrossRef]
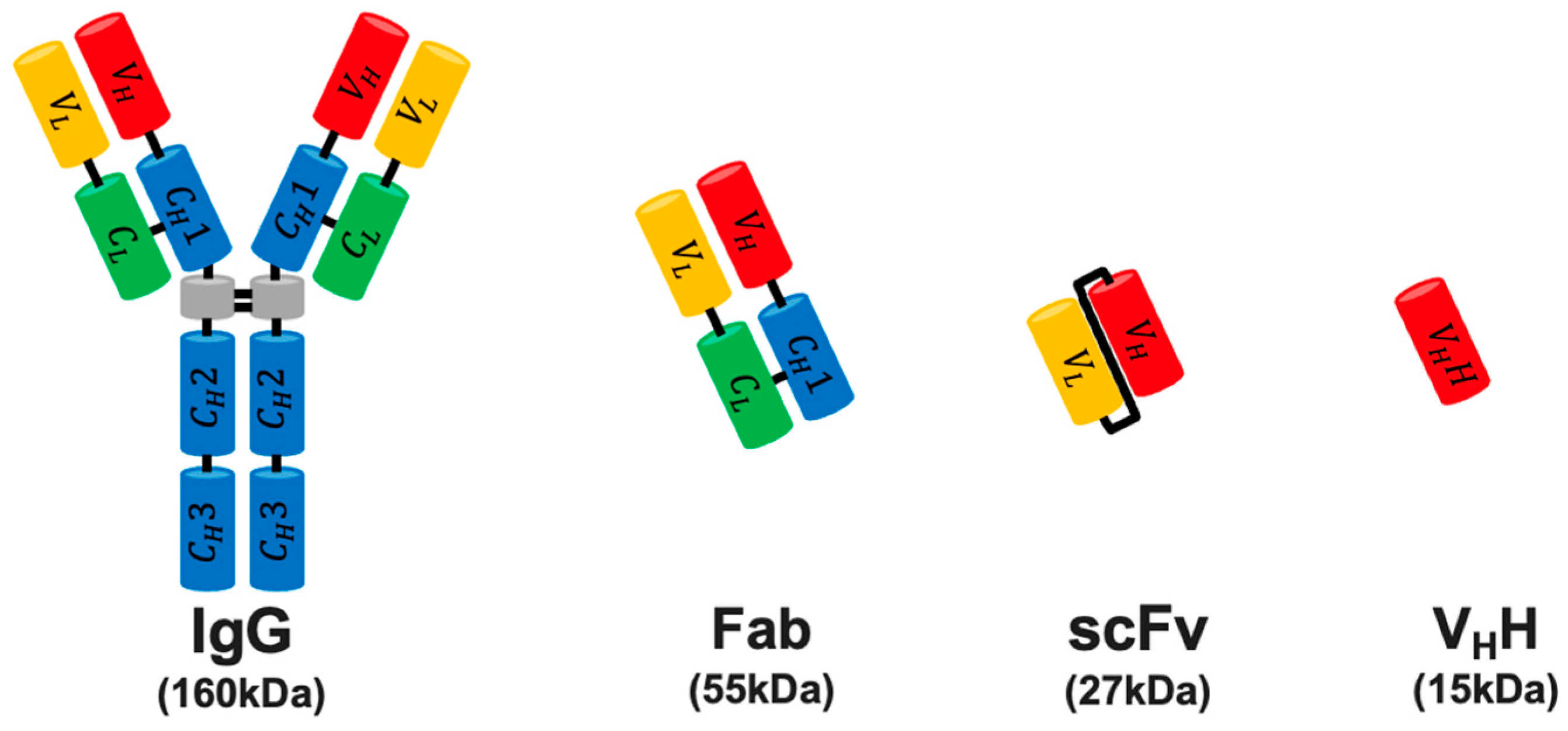
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.-M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-Chain Antigen-Binding Proteins. Science 1988, 242, 423–426. [Google Scholar] [CrossRef]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. scFv antibody: Principles and clinical application. J. Immunol. Res. 2012, 2012, 980250. [Google Scholar] [CrossRef]
- Baylet, A.; Vyumvuhore, R.; Laclaverie, M.; Marchand, L.; Mainzer, C.; Bordes, S.; Closs-Gonthier, B.; Delpy, L. Transcutaneous penetration of a single-chain variable fragment (scFv) compared to a full-size antibody: Potential tool for atopic dermatitis (AD) treatment. Allergy Asthma Clin. Immunol. 2021, 17, 73. [Google Scholar] [CrossRef]
- Duan, Y.; Chen, R.; Huang, Y.; Meng, X.; Chen, J.; Liao, C.; Tang, Y.; Zhou, C.; Gao, X.; Sun, J. Tuning the ignition of CAR: Optimizing the affinity of scFv to improve CAR-T therapy. Cell. Mol. Life Sci. 2021, 79, 14. [Google Scholar] [CrossRef]
- KÖHler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Schladetsch, M.A.; Wiemer, A.J. Generation of Single-Chain Variable Fragment (scFv) Libraries for Use in Phage Display. Curr. Protoc. 2021, 1, e182. [Google Scholar] [CrossRef]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef]
- Ling, W.-L.; Lua, W.-H.; Gan, S.K.-E. Sagacity in antibody humanization for therapeutics, diagnostics and research purposes: Considerations of antibody elements and their roles. Antib. Ther. 2020, 3, 71–79. [Google Scholar] [CrossRef]
- Hoogenboom, H.R. Selecting and screening recombinant antibody libraries. Nat. Biotechnol. 2005, 23, 1105–1116. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, Y.-L.; Lu, G.; Ji, H.; Rodi, C.P. Recombinant antibody libraries and selection technologies. New Biotechnol. 2011, 28, 448–452. [Google Scholar] [CrossRef]
- Pelat, T.; Thullier, P. Non-human primate immune libraries combined with germline humanization. mAbs 2009, 1, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Begent, R.H.J.; Verhaar, M.J.; Chester, K.A.; Casey, J.L.; Green, A.J.; Napier, M.P.; Hopestone, L.D.; Cushen, N.; Keep, P.A.; Johnson, C.J.; et al. Clinical evidence of efficient tumor targetting based on single–chain Fv antibody selected from a combinatorial library. Nat. Med. 1996, 2, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Buhr, D.; Gunter, C.; Frenette, J.; Ferguson, M.; Sanford, E.; Holland, E.; Rajagopal, C.; Batonick, M.; Kiss, M.M.; et al. Rational library design by functional CDR resampling. New Biotechnol. 2018, 45, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lerner, R.A. Combinatorial antibody libraries: New advances, new immunological insights. Nat. Rev. Immunol. 2016, 16, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, M.; Bu, W.; Joyce, M.G.; Meng, G.; Whittle, J.R.; Baxa, U.; Yamamoto, T.; Narpala, S.; Todd, J.P.; Rao, S.S.; et al. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 2015, 162, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Bazan, J.; Całkosiński, I.; Gamian, A. Phage display—A powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccin. Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef]
- Rader, C.; Barbas, C.F. Phage display of combinatorial antibody libraries. Curr. Opin. Biotechnol. 1997, 8, 503–508. [Google Scholar] [CrossRef]
- Rader, C. Generation of human Fab libraries for phage display. Methods Mol. Biol. 2012, 901, 53–79. [Google Scholar] [CrossRef]
- Barbas, C.F. Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Publishing Inc: New York, NY, USA, 2001. [Google Scholar]
- Wang, S.; Zheng, C.; Liu, Y.; Zheng, H.; Wang, Z. Construction of multiform scFv antibodies using linker peptide. J. Genet. Genom. 2008, 35, 313–316. [Google Scholar] [CrossRef]
- Holliger, P.; Prospero, T.; Winter, G. “Diabodies”: Small bivalent and bispecific antibody fragments. Proc. Natl. Acad. Sci. USA 1993, 90, 6444–6448. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Jia, X.; Feng, J.; Shen, B.; Huang, Y.; Geng, S.; Sun, Y.; Wang, Y.; Li, Y.; Long, M. Molecular Modeling and Affinity Determination of scFv Antibody: Proper Linker Peptide Enhances Its Activity. Ann. Biomed. Eng. 2010, 38, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Pardon, E.; Laeremans, T.; Triest, S.; Rasmussen, S.G.F.; Wohlkönig, A.; Ruf, A.; Muyldermans, S.; Hol, W.G.J.; Kobilka, B.K.; Steyaert, J. A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 2014, 9, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Thermo Scientific Inc. Thermo Scientific Immunoassay Plate Guide. 2018. Available online: https://assets.thermofisher.com/TFS-Assets/LCD/Scientific-Resources/Immunoassay_Plate_Guide.pdf (accessed on 20 November 2023).
- Lee, C.M.Y.; Iorno, N.; Sierro, F.; Christ, D. Selection of human antibody fragments by phage display. Nat. Protoc. 2007, 2, 3001–3008. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heavy Chain (VH) | Light Chain (VL) | ||
|---|---|---|---|
| κ Light | λ Light | ||
| V segment | 40 | 40 | 30 |
| D segment | 25 | - | - |
| J segment | 6 | 5 | 4 |
| Reaction Mixture | Touch Down PCR Condition | ||
|---|---|---|---|
| Platinum Hot Start Taq 2× Master Mix | 12.5 μL | 94 °C | 2 min |
| Autoclaved H2O | 7 μL | 94 °C | 30 s β |
| First-strand cDNA | 0.5 μL | 80 °C to 68 °C | 30 s (−1 °C/cycle) β |
| V region forward primer (10 μM) (V segment) α | 2.5 μL | 72 °C | 1.5 min β |
| V region reverse primer (10 μM) (J segment) α | 2.5 μL | 94 °C | 30 s χ |
| 50 °C | 30 s χ | ||
| 72 °C | 1.5 min χ | ||
| 72 °C | 10 min | ||
| 105 °C | Lid temp | ||
| Reaction Mixture | Touch Down PCR Condition | ||
|---|---|---|---|
| Platinum Hot Start Taq 2× Master Mix | 12.5 μL | 94 °C | 2 min |
| VH-short or VH-long | 50 ng | 94 °C | 30 s β |
| Vκ or Vλ | 50 ng | 56 °C | 30 s β |
| SOE primer—F α | 2.5 μL | 72 °C | 30 s β, 20 cycles |
| 12 °C χ | ∞ | ||
| SOE primer—R α | 2.5 μL | 94 °C | 30 s δ |
| Autoclaved H2O | To 25 μL | 68 °C | 30 s δ |
| 72 °C | 1 min δ | ||
| 72 °C | 10 min | ||
| 105 °C | Lid temp | ||
| Reaction Mixture | Control 1 (No Ligase) | Control 2 (No Insert) | Control 3 (with Stuffer Insert) | scFv | T4 DNA Ligase Reaction Condition | |
|---|---|---|---|---|---|---|
| T4 10× ligation buffer | 2 μL | 2 μL | 2 μL | 2 μL | 16 °C | 16 h |
| Digested scFv | - | - | - | 70 ng | 65 °C | 10 min |
| Stuffer fragment | - | - | 70 ng | - | 4 °C | Hold |
| Digested pComb3XSS | 140 ng | 140 ng | 140 ng | 140 ng | 105 °C | Lid temp |
| T4 ligase | - | 1 μL (400 U) | 1 μL (400 U) | 1 μL (400 U) | ||
| Autoclaved H2O | To 20 μL | To 20 μL | To 20 μL | To 20 μL | ||
| Sample ID | Heavy Chain V Segment Family | Heavy Chain J Segment Family | Light Chain V Segment Family | Light Chain J Segment Family | Sample ID | Heavy Chain V Segment Family | Heavy Chain J Segment Family | Light Chain V Segment Family | Light Chain J Segment Family | Sample ID | Heavy Chain V Segment Family | Heavy Chain J Segment Family | Light Chain V Segment Family | Light Chain J Segment Family |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QC1 | HUVH3E | Hujh3-S | HUVK6B | hujk1 | QC11 | HUVH6 | Hujh6b-L | HUVK1C | hujk2 | QC21 | HUVH3C | hujh45 | HUVL2C | hujl23 |
| QC2 | HUVH3B | Hujh45-S | HUVK3B | hujk2 | QC12 | HUVH1A | Hujh2-L | HUVL3C | hujl23 | QC22 | HUVH1A | hujh6b | HUVL2B | hujl23 |
| QC3 | HUVH1A | Hujh2-S | HUVL2B | hujl1 | QC13 | HUVH1C | Hujh6b-L | HUVK3C | hukj2 | QC23 | HUVH4C | hujh45 | HUVK1C | hujk2 |
| QC4 | HUVH3C | Hujh1-S | HUVL6 | hujl1 | QC14 | HUVH4A | Hujh3-S | HUVK3A | hujk4 | QC24 | HUVH1A | hujh45 | HUVK1B | hujk3 |
| QC5 | HUVH1C | Hujh2-L | HUVK1B | hujk1 | QC15 | HUVH1C | Hujh1-L | HUVK3A | hujk2 | QC25 | HUVH1A | Hujh1 | HUVL2C | hujl1 |
| QC6 | HUVH1A | Hujh3-L | HUVL5A9 | hujl1 | QC16 | HUVH4C | Hujh2-L | HUVK1A | hujk4 | QC26 | HUVH3E | Hujh3 | HUVL8 | hujl23 |
| QC7 | HUVH1C | Hujh3-S | HUVK2A | hujk1 | QC17 | HUVH4D | Hujh2-S | HUVL1A | hujl1 | QC27 | HUVH1C | Hujh3 | HUVL6 | hujl1 |
| QC8 | HUVH1C | Hujh45-L | HUVK3B | hujk3 | QC18 | HUVH4A | Hujh6a-S | HUVK2B | hujk1 | QC28 | HUVH4B | hujh6b | HUVL1B | hujl23 |
| QC9 | HUVH4D | Hujh45-L | HUVL10 | hujl1 | QC19 | HUVH4B | Hujh6a-S | HUVL2A | hujl7 | QC29 | HUVH2A | Hujh1 | HUVK1A | hujk3 |
| QC10 | HUVH1B | Hujh45-S | HUVL10 | hujl1 | QC20 | HUVH1A | hujh2 | HUVK6A | hujk4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Kim, D.; Kang, S.; Jung, J.U. A Detailed Protocol for Constructing a Human Single-Chain Variable Fragment (scFv) Library and Downstream Screening via Phage Display. Methods Protoc. 2024, 7, 13. https://doi.org/10.3390/mps7010013
Liu Z, Kim D, Kang S, Jung JU. A Detailed Protocol for Constructing a Human Single-Chain Variable Fragment (scFv) Library and Downstream Screening via Phage Display. Methods and Protocols. 2024; 7(1):13. https://doi.org/10.3390/mps7010013
Chicago/Turabian StyleLiu, Ziyi, Dokyun Kim, Seokmin Kang, and Jae U. Jung. 2024. "A Detailed Protocol for Constructing a Human Single-Chain Variable Fragment (scFv) Library and Downstream Screening via Phage Display" Methods and Protocols 7, no. 1: 13. https://doi.org/10.3390/mps7010013