
Newborn Screening for Fabry Disease: Current Status of Knowledge
 , ,
, ,
Abstract
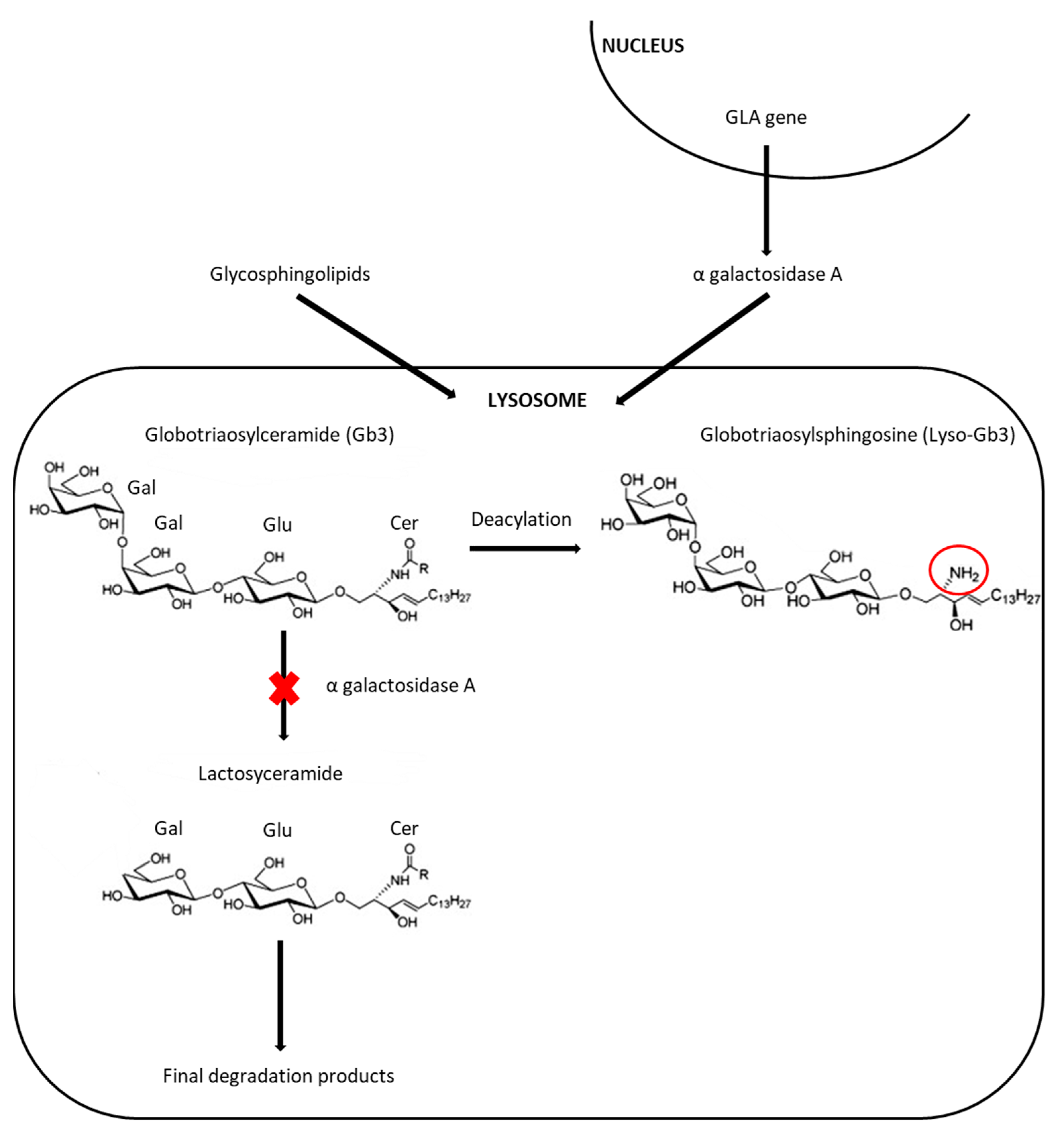
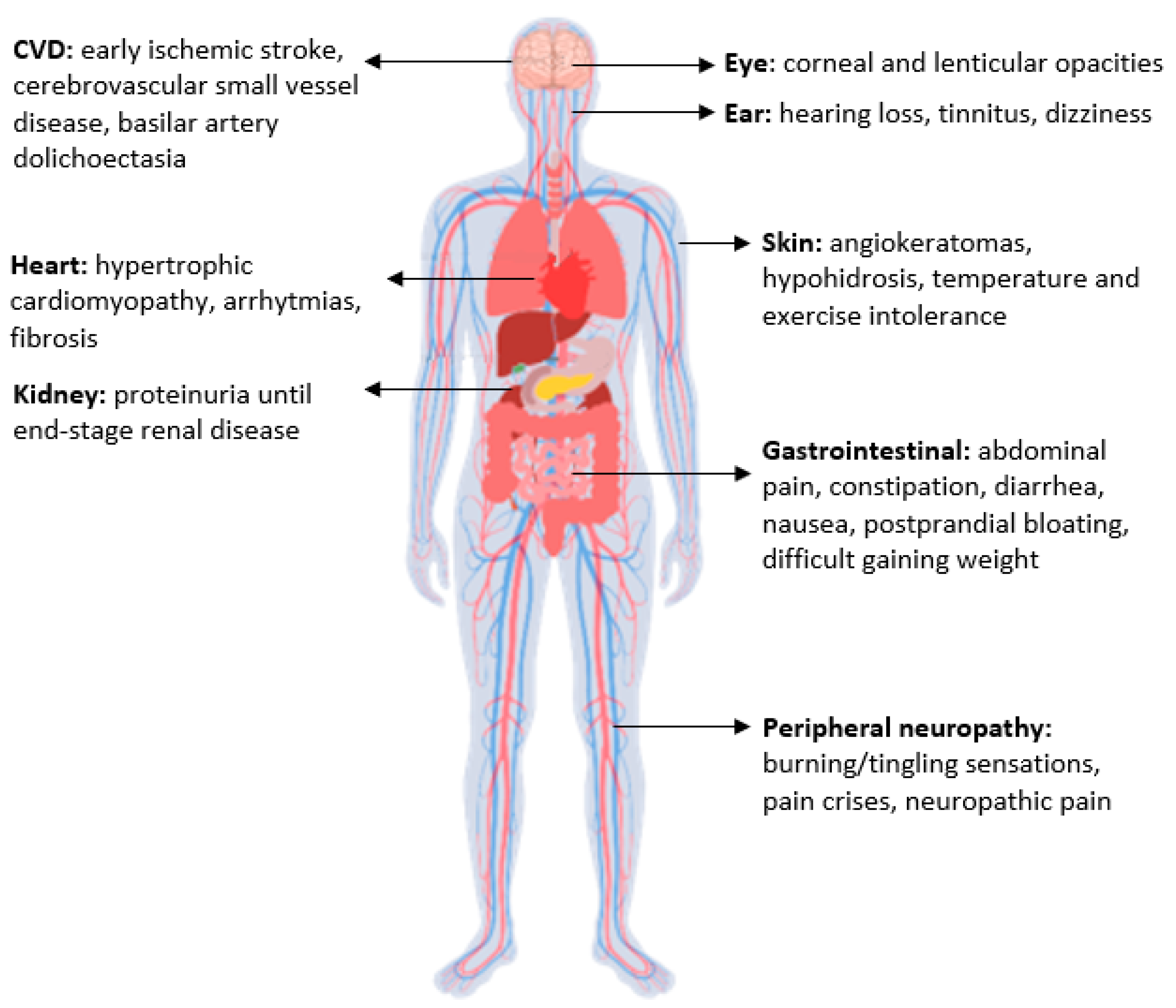
:1. Introduction
2. Methods
3. Results
3.1. Screening Methods
3.2. Second Tier Test
3.3. Genetic Screening
3.4. Newborn Screening for FD in the World
3.5. Recommendations for Management of Positive Neonates
3.6. Benefits and Challenges of FD Newborn Screening
4. Conclusions and Future Directions
- The lack of a second-tier test suitable to cover all the forms of the disease and reduce the recall rate;
- No biochemical detection of heterozygous females;
- The clinical interpretation of unclassified variants and VUS;
- The impact of early diagnosis on patients with later onset forms.
Funding
Conflicts of Interest
Abbreviations
References
- Germain, D.P. Fabry Disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, K.; Zwiers, K.C.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.M.F.G.; Artola, M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Laney, D.A.; Peck, D.S.; Atherton, A.M.; Manwaring, L.P.; Christensen, K.M.; Shankar, S.P.; Grange, D.K.; Wilcox, W.R.; Hopkin, R.J. Fabry disease in infancy and early childhood: A systematic literature review. Genet. Med. 2015, 17, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burlina, A.P.; Politei, J. Fabry Disease. In Neurometabolic Hereditary Diseases of Adults; Burlina, A.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 67–98. [Google Scholar]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. Alpha-Galactosidase A Deficiency: Fabry Disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Beck, M.; Cox, T.M. Comment: Why are females with Fabry disease affected? Mol. Genet. Metab. Rep. 2019, 21, 100529. [Google Scholar] [CrossRef]
- Massaccesi, L.; Burlina, A.; Baquero, C.J.; Goi, G.; Burlina, A.P.; Tettamanti, G. Whole-Blood Alpha-D-Galactosidase A Activity for the Identification of Fabry’s Patients. Clin. Biochem. 2011, 44, 916–921. [Google Scholar] [CrossRef]
- Effraimidis, G.; Feldt-Rasmussen, U.; Rasmussen, Å.K.; Lavoie, P.; Abaoui, M.; Boutin, M.; Auray-Blais, C. Globotriaosylsphingosine (Lyso-Gb 3) and Analogues in Plasma and Urine of Patients with Fabry Disease and Correlations with Long-Term Treatment and Genotypes in a Nationwide Female Danish Cohort. J. Med. Genet. 2021, 58, 692–700. [Google Scholar] [CrossRef]
- Schiffmann, R.; Murray, G.J.; Treco, D.; Daniel, P.; Sellos-Moura, M.; Myers, M.; Quirk, J.M.; Zirzow, G.C.; Borowski, M.; Loveday, K.; et al. Infusion of α-Galactosidase A Reduces Tissue Globotriaosylceramide Storage in Patients with Fabry Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase A Replacement Therapy in Fabry’s Disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Chong, K.-W.; Hsu, J.-H.; Yu, H.-C.; Shih, C.-C.; Huang, C.-H.; Lin, S.-J.; Chen, C.-H.; Chiang, C.-C.; Ho, H.-J.; et al. High Incidence of the Cardiac Variant of Fabry Disease Revealed by Newborn Screening in the Taiwan Chinese Population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkin, R.J.; Jefferies, J.L.; Laney, D.A.; Lawson, V.H.; Mauer, M.; Taylor, M.R.; Wilcox, W.R.; Fabry Pediatric Expert Panel. The management and treatment of children with Fabry disease: A United States-based perspective. Mol. Genet. Metab. 2016, 117, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Biegstraaten, M.; Arngrímsson, R.; Barbey, F.; Boks, L.; Cecchi, F.; Deegan, P.B.; Feldt-Rasmussen, U.; Geberhiwot, T.; Germain, D.P.; Hendriksz, C.; et al. Recommendations for Initiation and Cessation of Enzyme Replacement Therapy in Patients with Fabry Disease: The European Fabry Working Group Consensus Document. Orphanet J. Rare Dis. 2015, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oqvist, B.; Brenner, B.M.; Oliveira, J.P.; Ortiz, A.; Schaefer, R.; Svarstad, E.; Wanner, C.; Zhang, K.; Warnock, D.G. Nephropathy in Fabry Disease: The Importance of Early Diagnosis and Testing in High-Risk Populations. Nephrol. Dial. Transplant. 2009, 24, 1736–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.-R.; Hung, S.-C.; Chang, F.-P.; Yu, W.-C.; Sung, S.-H.; Hsu, C.-L.; Dzhagalov, I.; Yang, C.-F.; Chu, T.-H.; Lee, H.-J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence. J. Am. Coll. Cardiol. 2016, 68, 2554–2563. [Google Scholar] [CrossRef]
- Germain, D.P.; Altarescu, G.; Barriales-Villa, R.; Mignani, R.; Pawlaczyk, K.; Pieruzzi, F.; Terryn, W.; Vujkovac, B.; Ortiz, A. An Expert Consensus on Practical Clinical Recommendations and Guidance for Patients with Classic Fabry Disease. Mol. Genet. Metab. 2022, 137, 49–61. [Google Scholar] [CrossRef]
- Meikle, P.J.; Grasby, D.J.; Dean, C.J.; Lang, D.L.; Bockmann, M.; Whittle, A.M.; Fietz, M.J.; Simonsen, H.; Fuller, M.; Brooks, D.A.; et al. Newborn Screening for Lysosomal Storage Disorders. Mol. Genet. Metab. 2006, 88, 307–314. [Google Scholar] [CrossRef]
- Meikle, P.J.; Ranieri, E.; Simonsen, H.; Rozaklis, T.; Ramsay, S.L.; Whitfield, P.D.; Fuller, M.; Christensen, E.; Skovby, F.; Hopwood, J.J. Newborn Screening for Lysosomal Storage Disorders: Clinical Evaluation of a Two-Tier Strategy. Pediatrics 2004, 114, 909–916. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening*. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Hwu, W.-L.; Chien, Y.-H.; Lee, N.-C.; Chiang, S.-C.; Dobrovolny, R.; Huang, A.-C.; Yeh, H.-Y.; Chao, M.-C.; Lin, S.-J.; Kitagawa, T.; et al. Newborn Screening for Fabry Disease in Taiwan Reveals a High Incidence of the Later-Onset GLA Mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sista, R.S.; Eckhardt, A.E.; Wang, T.; Graham, C.; Rouse, J.L.; Norton, S.M.; Srinivasan, V.; Pollack, M.G.; Tolun, A.A.; Bali, D.; et al. Digital Microfluidic Platform for Multiplexing Enzyme Assays: Implications for Lysosomal Storage Disease Screening in Newborns. Clin. Chem. 2011, 57, 1444–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sista, R.; Eckhardt, A.E.; Wang, T.; Séllos-Moura, M.; Pamula, V.K. Rapid, Single-Step Assay for Hunter Syndrome in Dried Blood Spots Using Digital Microfluidics. Clin. Chim. Acta 2011, 412, 1895–1897. [Google Scholar] [CrossRef]
- Sista, R.S.; Wang, T.; Wu, N.; Graham, C.; Eckhardt, A.; Winger, T.; Srinivasan, V.; Bali, D.; Millington, D.S.; Pamula, V.K. Multiplex Newborn Screening for Pompe, Fabry, Hunter, Gaucher, and Hurler Diseases Using a Digital Microfluidic Platform. Clin. Chim. Acta 2013, 424, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Millington, D.; Bali, D. Current State of the Art of Newborn Screening for Lysosomal Storage Disorders. Int. J. Neonatal Screen. 2018, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Matern, D.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Tortorelli, S. Newborn Screening for Lysosomal Storage Disorders. Semin. Perinatol. 2015, 39, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Scott, C.R.; Chamoles, N.A.; Ghavami, A.; Pinto, B.M.; Turecek, F.; Gelb, M.H. Direct Multiplex Assay of Lysosomal Enzymes in Dried Blood Spots for Newborn Screening. Clin. Chem. 2004, 50, 1785–1796. [Google Scholar] [CrossRef] [Green Version]
- Duffey, T.A.; Bellamy, G.; Elliott, S.; Fox, A.C.; Glass, M.; Turecek, F.; Gelb, M.H.; Scott, C.R. A Tandem Mass Spectrometry Triplex Assay for the Detection of Fabry, Pompe, and Mucopolysaccharidosis-I (Hurler). Clin. Chem. 2010, 56, 1854–1861. [Google Scholar] [CrossRef] [Green Version]
- Gelb, M.H.; Scott, C.R.; Turecek, F. Newborn Screening for Lysosomal Storage Diseases. Clin. Chem. 2015, 61, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Gelb, M.; Lukacs, Z.; Ranieri, E.; Schielen, P. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2018, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, K.A.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Tortorelli, S.; Hopwood, J.J.; Lorey, F.; Majumdar, R.; Kroll, C.A.; McDonald, A.M.; et al. A Comparative Effectiveness Study of Newborn Screening Methods for Four Lysosomal Storage Disorders. Int. J. Neonatal Screen. 2020, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Spáčil, Z.; Elliott, S.; Reeber, S.L.; Gelb, M.H.; Scott, C.R.; Tureček, F. Comparative Triplex Tandem Mass Spectrometry Assays of Lysosomal Enzyme Activities in Dried Blood Spots Using Fast Liquid Chromatography: Application to Newborn Screening of Pompe, Fabry, and Hurler Diseases. Anal. Chem. 2011, 83, 4822–4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schielen, P.; Kemper, E.; Gelb, M. Newborn Screening for Lysosomal Storage Diseases: A Concise Review of the Literature on Screening Methods, Therapeutic Possibilities and Regional Programs. Int. J. Neonatal Screen. 2017, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Peake, R.; Bodamer, O. Newborn Screening for Lysosomal Storage Disorders. J. Pediatr. Genet. 2016, 6, 051–060. [Google Scholar] [CrossRef] [Green Version]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot Study of Newborn Screening for Six Lysosomal Storage Diseases Using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef]
- Kumar, A.B.; Masi, S.; Ghomashchi, F.; Chennamaneni, N.K.; Ito, M.; Scott, C.R.; Turecek, F.; Gelb, M.H.; Spacil, Z. Tandem Mass Spectrometry Has a Larger Analytical Range than Fluorescence Assays of Lysosomal Enzymes: Application to Newborn Screening and Diagnosis of Mucopolysaccharidoses Types II, IVA, and VI. Clin. Chem. 2015, 61, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Gelb, M.H.; Ronald Scott, C.; Turecek, F.; Liao, H.-C. Comparison of Tandem Mass Spectrometry to Fluorimetry for Newborn Screening of LSDs. Mol. Genet. Metab. Rep. 2017, 12, 80–81. [Google Scholar] [CrossRef]
- Liao, H.-C.; Chiang, C.-C.; Niu, D.-M.; Wang, C.-H.; Kao, S.-M.; Tsai, F.-J.; Huang, Y.-H.; Liu, H.-C.; Huang, C.-K.; Gao, H.-J.; et al. Detecting Multiple Lysosomal Storage Diseases by Tandem Mass Spectrometry—A National Newborn Screening Program in Taiwan. Clin. Chim. Acta 2014, 431, 80–86. [Google Scholar] [CrossRef]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef] [Green Version]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and Dried Blood Spot Lysosphingolipids for the Diagnosis of Different Sphingolipidoses: A Comparative Study. Clin. Chem. Lab. Med. CCLM 2019, 57, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Malvagia, S.; Ferri, L.; Della Bona, M.; Borsini, W.; Cirami, C.L.; Dervishi, E.; Feriozzi, S.; Gasperini, S.; Motta, S.; Mignani, R.; et al. Multicenter Evaluation of Use of Dried Blood Spot Compared to Conventional Plasma in Measurements of Globotriaosylsphingosine (LysoGb3) Concentration in 104 Fabry Patients. Clin. Chem. Lab. Med. CCLM 2021, 59, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Gragnaniello, V.; Burlina, A.P.; Polo, G.; Giuliani, A.; Salviati, L.; Duro, G.; Cazzorla, C.; Rubert, L.; Maines, E.; Germain, D.P.; et al. Newborn Screening for Fabry Disease in Northeastern Italy: Results of Five Years of Experience. Biomolecules 2021, 11, 951. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Lee, N.-C.; Chen, P.-W.; Yeh, H.-Y.; Gelb, M.H.; Chiu, P.-C.; Chu, S.-Y.; Lee, C.-H.; Lee, A.-R.; Hwu, W.-L. Newborn Screening for Morquio Disease and Other Lysosomal Storage Diseases: Results from the 8-Plex Assay for 70,000 Newborns. Orphanet J. Rare Dis. 2020, 15, 38. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Kasper, D.; Pagliardini, V.; Biamino, E.; Giachero, S.; Porta, F. Metabolic Progression to Clinical Phenotype in Classic Fabry Disease. Ital. J. Pediatr. 2017, 43, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kritzer, A.; Siddharth, A.; Leestma, K.; Bodamer, O. Early Initiation of Enzyme Replacement Therapy in Classical Fabry Disease Normalizes Biomarkers in Clinically Asymptomatic Pediatric Patients. Mol. Genet. Metab. Rep. 2019, 21, 100530. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.-L.; Liu, M.-Y.; Yu, H.-C.; Chiang, C.-C.; Chiang, H.; Suen, J.-H.; Kao, S.-M.; Huang, Y.-H.; Wu, T.J.-T.; Yang, C.-F.; et al. The Use of High Resolution Melting Analysis to Detect Fabry Mutations in Heterozygous Females via Dry Bloodspots. Clin. Chim. Acta 2012, 413, 422–427. [Google Scholar] [CrossRef]
- Lee, S.-H.; Li, C.-F.; Lin, H.-Y.; Lin, C.-H.; Liu, H.-C.; Tsai, S.-F.; Niu, D.-M. High-Throughput Detection of Common Sequence Variations of Fabry Disease in Taiwan Using DNA Mass Spectrometry. Mol. Genet. Metab. 2014, 111, 507–512. [Google Scholar] [CrossRef]
- Lu, Y.-H.; Huang, P.-H.; Wang, L.-Y.; Hsu, T.-R.; Li, H.-Y.; Lee, P.-C.; Hsieh, Y.-P.; Hung, S.-C.; Wang, Y.-C.; Chang, S.-K.; et al. Improvement in the Sensitivity of Newborn Screening for Fabry Disease among Females through the Use of a High-Throughput and Cost-Effective Method, DNA Mass Spectrometry. J. Hum. Genet. 2018, 63, 1–8. [Google Scholar] [CrossRef]
- Colon, C.; Ortolano, S.; Melcon-Crespo, C.; Alvarez, J.V.; Lopez-Suarez, O.E.; Couce, M.L.; Fernández-Lorenzo, J.R. Newborn Screening for Fabry Disease in the North-West of Spain. Eur. J. Pediatr. 2017, 176, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Paciotti, S.; Persichetti, E.; Pagliardini, S.; Deganuto, M.; Rosano, C.; Balducci, C.; Codini, M.; Filocamo, M.; Menghini, A.R.; Pagliardini, V.; et al. First Pilot Newborn Screening for Four Lysosomal Storage Diseases in an Italian Region: Identification and Analysis of a Putative Causative Mutation in the GBA Gene. Clin. Chim. Acta 2012, 413, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; De Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal Screening for Lysosomal Storage Disorders: Feasibility and Incidence from a Nationwide Study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Wittmann, J.; Karg, E.; Turi, S.; Legnini, E.; Wittmann, G.; Giese, A.-K.; Lukas, J.; Gölnitz, U.; Klingenhäger, M.; Bodamer, O.; et al. Newborn Screening for Lysosomal Storage Disorders in Hungary. In JIMD Reports—Case and Research Reports, 2012/3; SSIEM, Ed.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2012; Volume 6, pp. 117–125. [Google Scholar] [CrossRef] [Green Version]
- Sawada, T.; Kido, J.; Yoshida, S.; Sugawara, K.; Momosaki, K.; Inoue, T.; Tajima, G.; Sawada, H.; Mastumoto, S.; Endo, F.; et al. Newborn Screening for Fabry Disease in the Western Region of Japan. Mol. Genet. Metab. Rep. 2020, 22, 100562. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Hattori, K.; Ihara, K.; Ishii, A.; Nakamura, K.; Hirose, S. Newborn Screening for Fabry Disease in Japan: Prevalence and Genotypes of Fabry Disease in a Pilot Study. J. Hum. Genet. 2013, 58, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinen, Y.; Nakamura, S.; Yoshida, T.; Maruyama, H.; Nakamura, K. A New Mutation Found in Newborn Screening for Fabry Disease Evaluated by Plasma Globotriaosylsphingosine Levels. Hum. Genome Var. 2017, 4, 17002. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-C.; Hsu, T.-R.; Young, L.; Chiang, C.-C.; Huang, C.-K.; Liu, H.-C.; Niu, D.-M.; Chen, Y.-J. Functional and Biological Studies of α-Galactosidase A Variants with Uncertain Significance from Newborn Screening in Taiwan. Mol. Genet. Metab. 2018, 123, 140–147. [Google Scholar] [CrossRef]
- Chiang, S.-C.; Chen, P.-W.; Hwu, W.-L.; Lee, A.-J.; Chen, L.-C.; Lee, N.-C.; Chiou, L.-Y.; Chien, Y.-H. Performance of the Four-Plex Tandem Mass Spectrometry Lysosomal Storage Disease Newborn Screening Test: The Necessity of Adding a 2nd Tier Test for Pompe Disease. Int. J. Neonatal Screen. 2018, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Tian, L.; Gao, Q.; Guo, Y.; Li, G.; Li, Y.; Sun, M.; Yan, Y.; Li, Q.; Nie, W.; et al. Establishment of Cutoff Values for Newborn Screening of Six Lysosomal Storage Disorders by Tandem Mass Spectrometry. Front. Pediatr. 2022, 10, 814461. [Google Scholar] [CrossRef]
- Scott, C.R.; Elliott, S.; Buroker, N.; Thomas, L.I.; Keutzer, J.; Glass, M.; Gelb, M.H.; Turecek, F. Identification of Infants at Risk for Developing Fabry, Pompe, or Mucopolysaccharidosis-I from Newborn Blood Spots by Tandem Mass Spectrometry. J. Pediatr. 2013, 163, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York Pilot Newborn Screening Program for Lysosomal Storage Diseases: Report of the First 65,000 Infants. Genet. Med. 2019, 21, 631–640. [Google Scholar] [CrossRef]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Martínez, J.I.; Limón-Rojas, A.E.; Gaytán-García, M.D.J.; Reyna-Figueroa, J.; Wakida-Kusunoki, G.; Delgado-Calvillo, M.D.R.; Cantú-Reyna, C.; Cruz-Camino, H.; Cervantes-Barragán, D.E. Newborn Screening for Six Lysosomal Storage Disorders in a Cohort of Mexican Patients: Three-Year Findings from a Screening Program in a Closed Mexican Health System. Mol. Genet. Metab. 2017, 121, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Camargo Neto, E.; Schulte, J.; Pereira, J.; Bravo, H.; Sampaio-Filho, C.; Giugliani, R. Neonatal Screening for Four Lysosomal Storage Diseases with a Digital Microfluidics Platform: Initial Results in Brazil. Genet. Mol. Biol. 2018, 41, 414–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.-Q. Alternative Splicing in the A-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Previously Nominated Conditions. Available online: http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/nominatecondition/workgroup.html (accessed on 1 April 2023).
- Gelb, M. Newborn Screening for Lysosomal Storage Diseases: Methodologies, Screen Positive Rates, Normalization of Datasets, Second-Tier Tests, and Post-Analysis Tools. Int. J. Neonatal Screen. 2018, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn Screening for Lysosomal Storage Disorders by Tandem Mass Spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef]
- CLIR. Available online: https://Clir.Mayo.Edu/ (accessed on 1 April 2023).
- Hall, P.L.; Marquardt, G.; McHugh, D.M.S.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical Tools Improve Performance of Newborn Screening by Tandem Mass Spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.Y.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R. Lysosomal Storage Diseases: Diagnostic Confirmation and Management of Presymptomatic Individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Moiseev, S.; Suárez-Obando, F.; Al Ismaili, F.; Al Khawaja, H.; Altarescu, G.; Barreto, F.C.; Haddoum, F.; Hadipour, F.; Maksimova, I.; et al. The Benefits and Challenges of Family Genetic Testing in Rare Genetic Diseases—Lessons from Fabry Disease. Mol. Genet. Genom. Med. 2021, 9, e1666. [Google Scholar] [CrossRef]
- Laney, D.A.; Fernhoff, P.M. Diagnosis of Fabry Disease via Analysis of Family History. J. Genet. Couns. 2008, 17, 79–83. [Google Scholar] [CrossRef]
- Wilson, J.M.G.; Jungner, G.; WHO. Principles and Practice of Screening for Disease; Public Health Papers no. 34; World Health Organization: Geneva, Switzerland, 1968; 163p. [Google Scholar]
- Burlina, A.; Jones, S.A.; Chakrapani, A.; Church, H.J.; Heales, S.; Wu, T.H.Y.; Morton, G.; Roberts, P.; Sluys, E.F.; Cheillan, D. A New Approach to Objectively Evaluate Inherited Metabolic Diseases for Inclusion on Newborn Screening Programmes. Int. J. Neonatal Screen. 2022, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Cheillan, D.; Chakrapani, A.; Church, H.J.; Heales, S.; Wu, T.H.Y.; Morton, G.; Roberts, P.; Sluys, E.F.; Burlina, A. Application of a Novel Algorithm for Expanding Newborn Screening for Inherited Metabolic Disorders across Europe. Int. J. Neonatal Screen. 2022, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Kokotos, F. The Vulnerable Child Syndrome. Pediatr. Rev. 2009, 30, 193–194. [Google Scholar] [CrossRef]
- Timmermans, S.; Buchbinder, M. Patients-in-Waiting: Living between Sickness and Health in the Genomics Era. J. Health Soc. Behav. 2010, 51, 408–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, M.; Ohashi, T.; Fukuda, T.; Yanagisawa, T.; Inomata, T.; Nagaoka, T.; Kitagawa, T.; Eto, Y.; Ida, H.; Kusano, E. No Accumulation of Globotriaosylceramide in the Heart of a Patient with the E66Q Mutation in the α-Galactosidase A Gene. Mol. Genet. Metab. 2012, 107, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Togawa, T.; Tsukimura, T.; Kodama, T.; Tanaka, T.; Kawashima, I.; Saito, S.; Ohno, K.; Fukushige, T.; Kanekura, T.; Satomura, A.; et al. Fabry Disease: Biochemical, Pathological and Structural Studies of the α-Galactosidase A with E66Q Amino Acid Substitution. Mol. Genet. Metab. 2012, 105, 615–620. [Google Scholar] [CrossRef]
- Yasuda, M.; Shabbeer, J.; Benson, S.D.; Maire, I.; Burnett, R.M.; Desnick, R.J. Fabry Disease: Characterization of Alpha-Galactosidase A Double Mutations and the D313Y Plasma Enzyme Pseudodeficiency Allele. Hum. Mutat. 2003, 22, 486–492. [Google Scholar] [CrossRef]
- Eng, C.M.; Resnick-Silverman, L.A.; Niehaus, D.J.; Astrin, K.H.; Desnick, R.J. Nature and Frequency of Mutations in the A-Galactosidase A Gene That Cause Fabry Disease. Am. J. Hum. Genet. 1993, 53, 1186–1197. [Google Scholar]
- Viall, S.; Dennis, A.; Yang, A. Newborn Screening for Fabry Disease in Oregon: Approaching the Iceberg of A143T and Variants of Uncertain Significance. Am. J. Med. Genet. Part C 2022, 190, 206–214. [Google Scholar] [CrossRef]
- Elliott, P.; Baker, R.; Pasquale, F.; Quarta, G.; Ebrahim, H.; Mehta, A.B.; Hughes, D.A.; on behalf of the ACES Study Group. Prevalence of Anderson-Fabry Disease in Patients with Hypertrophic Cardiomyopathy: The European Anderson-Fabry Disease Survey. Heart 2011, 97, 1957–1960. [Google Scholar] [CrossRef]
- De Brabander, I.; Yperzeele, L.; Ceuterick-De Groote, C.; Brouns, R.; Baker, R.; Belachew, S.; Delbecq, J.; De Keulenaer, G.; Dethy, S.; Eyskens, F.; et al. Phenotypical Characterization of α-Galactosidase A Gene Mutations Identified in a Large Fabry Disease Screening Program in Stroke in the Young. Clin. Neurol. Neurosurg. 2013, 115, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Tholey, A.; Jakoby, T.; Vogelsberger, R.; Mönnikes, R.; Rossmann, H.; Beck, M.; Lackner, K.J. Quantification of the Fabry Marker LysoGb3 in Human Plasma by Tandem Mass Spectrometry. J. Chromatogr. B 2012, 883–884, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Terryn, W.; Vanholder, R.; Hemelsoet, D.; Leroy, B.P.; Van Biesen, W.; De Schoenmakere, G.; Wuyts, B.; Claes, K.; De Backer, J.; De Paepe, G.; et al. Questioning the Pathogenic Role of the GLA p.Ala143Thr “Mutation” in Fabry Disease: Implications for Screening Studies and ERT. In JIMD Reports—Case and Research Reports, 2012/5; Zschocke, J., Gibson, K.M., Brown, G., Morava, E., Peters, V., Eds.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2012; Volume 8, pp. 101–108. [Google Scholar] [CrossRef] [Green Version]
- Lenders, M.; Weidemann, F.; Kurschat, C.; Canaan-Kühl, S.; Duning, T.; Stypmann, J.; Schmitz, B.; Reiermann, S.; Krämer, J.; Blaschke, D.; et al. Alpha-Galactosidase A p.A143T, a Non-Fabry Disease-Causing Variant. Orphanet J. Rare Dis. 2016, 11, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GnomAD: The Genome Aggregation Database. Available online: https://Gnomad.Broadinstitute.Org (accessed on 1 April 2023).
- Garman, S.C. Structure-Function Relationships in α-Galactosidase A: Structure-Function Relationships in α-Galactosidase A. Acta Paediatr. 2007, 96, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P.; Levade, T.; Hachulla, E.; Knebelmann, B.; Lacombe, D.; Seguin, V.L.; Nguyen, K.; Noël, E.; Rabès, J. Challenging the Traditional Approach for Interpreting Genetic Variants: Lessons from Fabry Disease. Clin. Genet. 2022, 101, 390–402. [Google Scholar] [CrossRef]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M.F.G. Is It Fabry Disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef] [Green Version]
- Linthorst, G.E.; Poorthuis, B.J.H.M.; Hollak, C.E.M. Enzyme Activity for Determination of Presence of Fabry Disease in Women Results in 40% False-Negative Results. J. Am. Coll. Cardiol. 2008, 51, 2082. [Google Scholar] [CrossRef]
- Linthorst, G.E.; Vedder, A.C.; Aerts, J.M.F.G.; Hollak, C.E.M. Screening for Fabry Disease Using Whole Blood Spots Fails to Identify One-Third of Female Carriers. Clin. Chim. Acta 2005, 353, 201–203. [Google Scholar] [CrossRef]
- Hsu, T.-R.; Niu, D.-M. Fabry Disease: Review and Experience during Newborn Screening. Trends Cardiovasc. Med. 2018, 28, 274–281. [Google Scholar] [CrossRef]
- Burlina, A.; Brand, E.; Hughes, D.; Kantola, I.; Krämer, J.; Nowak, A.; Tøndel, C.; Wanner, C.; Spada, M. An expert consensus on the recommendations for the use of biomarkers in Fabry disease. Mol. Genet. Metab. 2023, 139, 107585. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, M.G.; de Ru, M.H.; Linthorst, G.E.; Hollak, C.E.M.; Wijburg, F.A.; van Zwieten, M.C.B. Fabry Patients’ Experiences with the Timing of Diagnosis Relevant for the Discussion on Newborn Screening. Mol. Genet. Metab. 2013, 109, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Lisi, E.C.; Ali, N. Opinions of Adults Affected with Later-onset Lysosomal Storage Diseases Regarding Newborn Screening: A Qualitative Study. J. Genet. Couns. 2021, 30, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Lisi, E.C.; Gillespie, S.; Laney, D.; Ali, N. Patients’ Perspectives on Newborn Screening for Later-Onset Lysosomal Storage Diseases. Mol. Genet. Metab. 2016, 119, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Lisi, E.C.; McCandless, S.E. Newborn Screening for Lysosomal Storage Disorders: Views of Genetic Healthcare Providers. J. Genet. Couns. 2016, 25, 373–384. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristics | Fluorometry | Digital Microfluidics | Tandem Mass Spectrometry | Immune Quantification |
|---|---|---|---|---|
| Method | enzymatic assay | enzymatic assay | enzymatic assay | protein abundance |
| Multiplexable | no | yes | yes | yes |
| Incubation time | overnight | 3 h | overnight | overnight |
| Assay conditions (specific pH, additives, buffers) | optimal | optimal | fixed pH (4.7) | not applicable |
| Interferences | low | low | very low | non-functioning enzyme |
| Analytical range | good | good | very good | not applicable |
| Instrumentation costs | low | low | high | low |
| Assay costs | low | intermediate | intermediate | low |
| Reagents | commercially available | commercially available | commercially available | not commercially available |
| Laboratory training | simple | simple | intermediate | Intermediate |
| Automation | Intermediate | high | high | Intermediate |
| Sample throughput | low | intermediate | high | low |
| Molecular Assay | Pros | Cons |
|---|---|---|
| High resolution melting | Cover the 7 exons and the IVS4 variant | Low sensitivity for variants located at exons 2 and 6 Sensitivity to variable concentrations of nucleic acid or salts Need of experience for periodic parameters adjustment Not reliable for males |
| Agena iPlex | Not stringent DNA quality control Easy, simple training Less than one day | Only known pathogenic variants |
| Study Period | Country | Method | Type of Cutoff | Number of NBS Samples | Number of below Cutoff Samples | Number of below Cutoff Samples/100,000 Newborns | Confirmed Patients from Genetic Analysis * | Presumed Incidence ** | Source of Data |
|---|---|---|---|---|---|---|---|---|---|
| Europe | |||||||||
| 2003–2005 | Italy | Fluorometric enzyme assay | fixed | 37,104 (only males) | 12 (m) | 32 (m) | 12 (m) | 1:3100 (m) | Spada et al. [22] |
| 2008 | Spain | Fluorometric enzyme assay | fixed | 14,600 (m 7575) | 106 (m 68) | 726 (m 898) | 37 (m 20) | 1:394 (m 1:378) | Colon et al. [52] |
| 2010–2012 | Italy | Fluorometric enzyme assay | fixed | 3403 (m 1702) | 0 | 0 | 0 | / | Paciotti et al. [53] |
| 2010 ** | Austria | MS/MS | fixed | 34,736 (deidentified) | 28 | 81 | 9 (m 6) | 1:3860 | Mechtler et al. [54] |
| 2011 *** | Hungary | MS/MS | fixed | 40,024 (deidentified) | 34 | 85 | 3 | 1:13,341 | Wittmann et al. [55] |
| 2015–2021 | Italy | MS/MS | fixed | 173,342 (m 89,485) | 23 (m 22) | 13 (m 25) | 22 (m) | 1:7879 (m 1:4068) | Gragnaniello et al. [45] |
| Asia | |||||||||
| 2006–2008 | Taiwan | Fluorometric enzyme assay | fixed | 171,977 (m 90,288) | 94 (m 91) | 55 (m 53) | 75 (m 73) | 1:2293 (m 1:1237) | Hwu et al. [23] |
| 2006–2018 | Japan | Fluorometric enzyme assay | fixed | 599,711 | 138 | 23 | 108 (m 64) | 1:5552 | Sawada et al. [56] |
| 2007–2010 | Japan | Fluorometric enzyme assay | fixed | 21,170 (m 10,827) | 7 (m 5) | 33 (m 46) | 6 (5 m) | 1:3024 (m 1:2166) | Inoue et al. [57] |
| 2007–2014 | Japan | Fluorometric enzyme assay | fixed | 2443 | 2 (m 2) | 82 | 2 (m 2) | 1:1222 | Chinen et al. [58] |
| 2008–2014 | Taiwan | Fluorometric enzyme assay then MS/MS | fixed | 792,247 (m 412,299) | 764 (m 425) | 96 (m 103) | 324 (m 272) | 1:2445 (m 1:1515) | Liao et al. [59] |
| 2010–2013 | Taiwan | MS/MS (compared with fluorometry) | fixed | 191,767 | 79 | 41 | 64 (m 61) | 1:2996 | Liao et al. [41] |
| 2015–2019 | Taiwan | MS/MS | fixed | 137,891 | 13 | 19 | 13 | 1:10,607 | Chiang et al. [60], Chien et al. [46] |
| 2019–2022 | China | MS/MS | %DMA | 38,945 | 21 | 54 | 3 | 1:12,982 | Li et al. [61] |
| USA | |||||||||
| 2011–2013 *** | California | MS/MS, immunocapture assay, DMF (comparative) | 89,508 (m 44,664) (deidentified) | Variable based on method | Not applicable | 50 (m 46) | 1:1790 (m 1:1970) | Sanders et al. [34] | |
| 2013 ** | Washington State | MS/MS | %DMA | 108,905 (m 54,800) (deidentified) | 16 (m 13) | 15 (m 24) | 7 (m 7) | 1:15,558 (m 1:7800) | Scott et al. [62] |
| 2013 | Missouri | DMF | fixed | 43,701 | 28 | 64 | 15 (m 15) | 1:2913 | Hopkins et al. [14] |
| 2013–2019 | New York | MS/MS | % DMA | 65,605 | 31 | 47 | 7 (m 7) | 1:9372 | Wasserstein et al. [63] |
| 2014–2016 | Illinois | MS/MS | % DMA | 219,793 | 107 | 49 | 32 (m 32) | 1:6968 | Burton et al. [64] |
| 2016 *** | Washington State | MS/MS | % DMA | 43,000 (deidentified) | 8 | 19 | 6 | 1:7167 | Elliot et al. [38] |
| Latin America | |||||||||
| 2012–2016 | Petroleos Mexicanos Health Services | MS/MS | fixed | 20,018 (m 10,241) | 5 (m 5) | 25 (m 49) | 5 (m 5) | 1:4003 (m 1:2048) | Navarrete-Martinez et al. [65] |
| 2017 | Brazil | DMF | fixed | 10,527 | 0 | 0 | 0 | / | Camargo Neto et al. [66] |
| Timing | Suggested Tests |
|---|---|
| Diagnostic confirmation | Genetic analysis * (patient and parents), substrate quantification (plasma lysoGb3) and enzyme activity in leukocytes, lymphocytes or plasma (in males). |
| Baseline diagnostic studies | ECG, echocardiogram, ophthalmologic examination, renal function tests, plasma and/or urine GL3 |
| Follow up every 6 months (classic form) or 12 months (later onset form) | Clinical examination (angiokeratomas, hypohidrosis, gastrointestinal symptoms, limb pain), kidney (eGFR according to Schwartz formula, microalbuminuria, proteinuria), cardiac assessments (ECG, echocardiography, 24-h holter), neurologic evaluation, plasma lyso-Gb3. |
| Advantages | Disadvantages |
|---|---|
| Available methods for NBS on DBS | Enzyme based assays do not identify many female heterozygotes. |
| Approved treatments | Higher than expected numbers of later onset forms |
| Importance of early diagnosis and treatment, often delayed clinical diagnosis | Lack of definite guidelines for follow up and start therapy especially for later onset forms |
| Better knowledge of the natural history | Frequent detection of VUS or benign variants |
| Genetic counseling | Phenotype prediction can be difficult |
| Family screening | |
| High incidence, more frequent than previously expected |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gragnaniello, V.; Burlina, A.P.; Commone, A.; Gueraldi, D.; Puma, A.; Porcù, E.; Stornaiuolo, M.; Cazzorla, C.; Burlina, A.B. Newborn Screening for Fabry Disease: Current Status of Knowledge. Int. J. Neonatal Screen. 2023, 9, 31. https://doi.org/10.3390/ijns9020031
Gragnaniello V, Burlina AP, Commone A, Gueraldi D, Puma A, Porcù E, Stornaiuolo M, Cazzorla C, Burlina AB. Newborn Screening for Fabry Disease: Current Status of Knowledge. International Journal of Neonatal Screening. 2023; 9(2):31. https://doi.org/10.3390/ijns9020031
Chicago/Turabian StyleGragnaniello, Vincenza, Alessandro P. Burlina, Anna Commone, Daniela Gueraldi, Andrea Puma, Elena Porcù, Maria Stornaiuolo, Chiara Cazzorla, and Alberto B. Burlina. 2023. "Newborn Screening for Fabry Disease: Current Status of Knowledge" International Journal of Neonatal Screening 9, no. 2: 31. https://doi.org/10.3390/ijns9020031