
Newborn Screening of Primary Carnitine Deficiency: An Overview of Worldwide Practices and Pitfalls to Define an Algorithm before Expansion of Newborn Screening in France
 , , , , , and
, , , , , and
Abstract
:1. Introduction
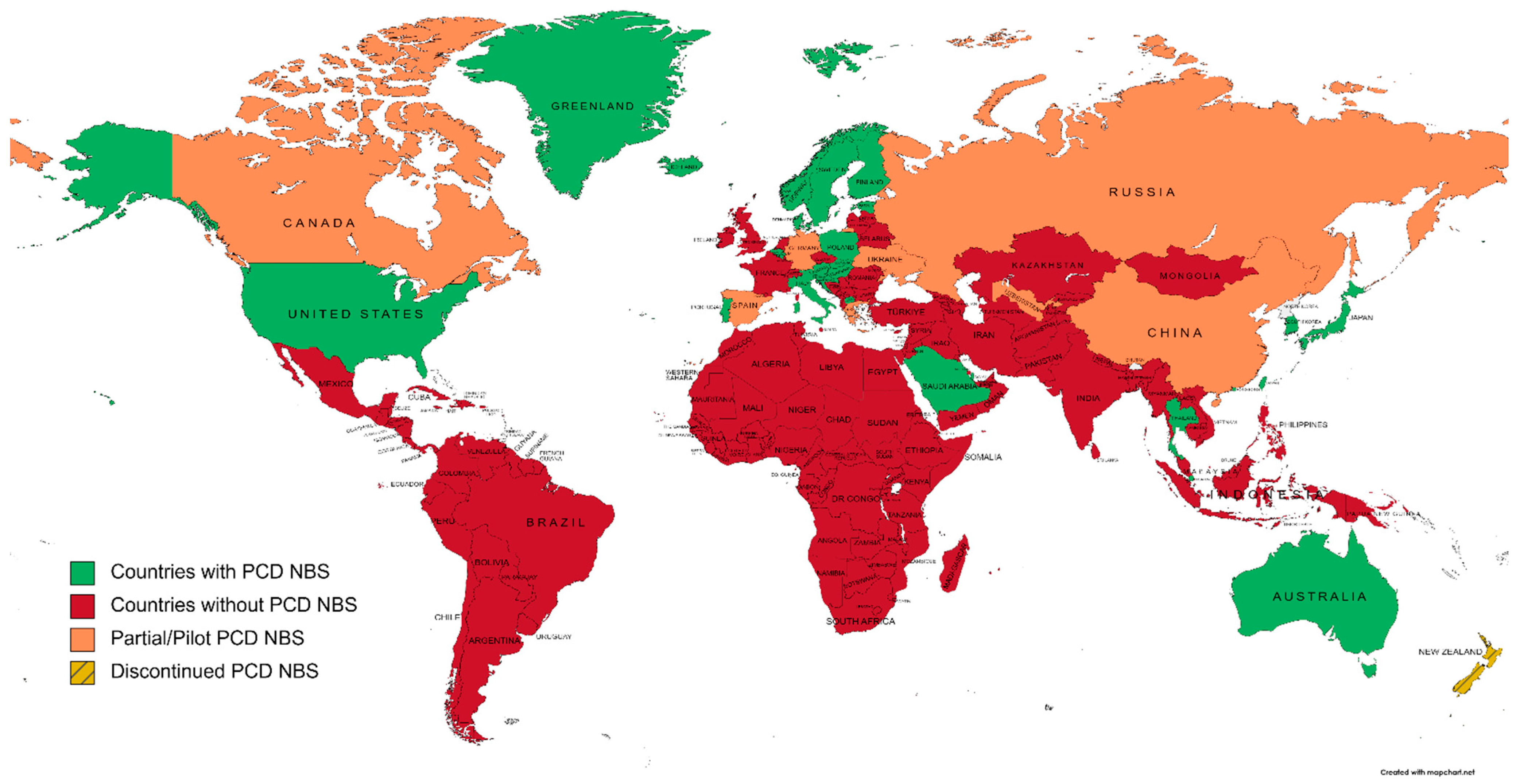
2. Worldwide Overview of Primary Carnitine Deficiency Newborn Screening
2.1. Countries/Regions Screening PCD
2.1.1. Australia and New Zealand
2.1.2. North America
2.1.3. Central and South America
2.1.4. Europe
2.1.5. Africa
2.1.6. Asia
2.1.7. Russia
3. Reports of NBS for PCD Worldwide
4. Molecular Findings
5. Pitfalls of Newborn Screening for PCD
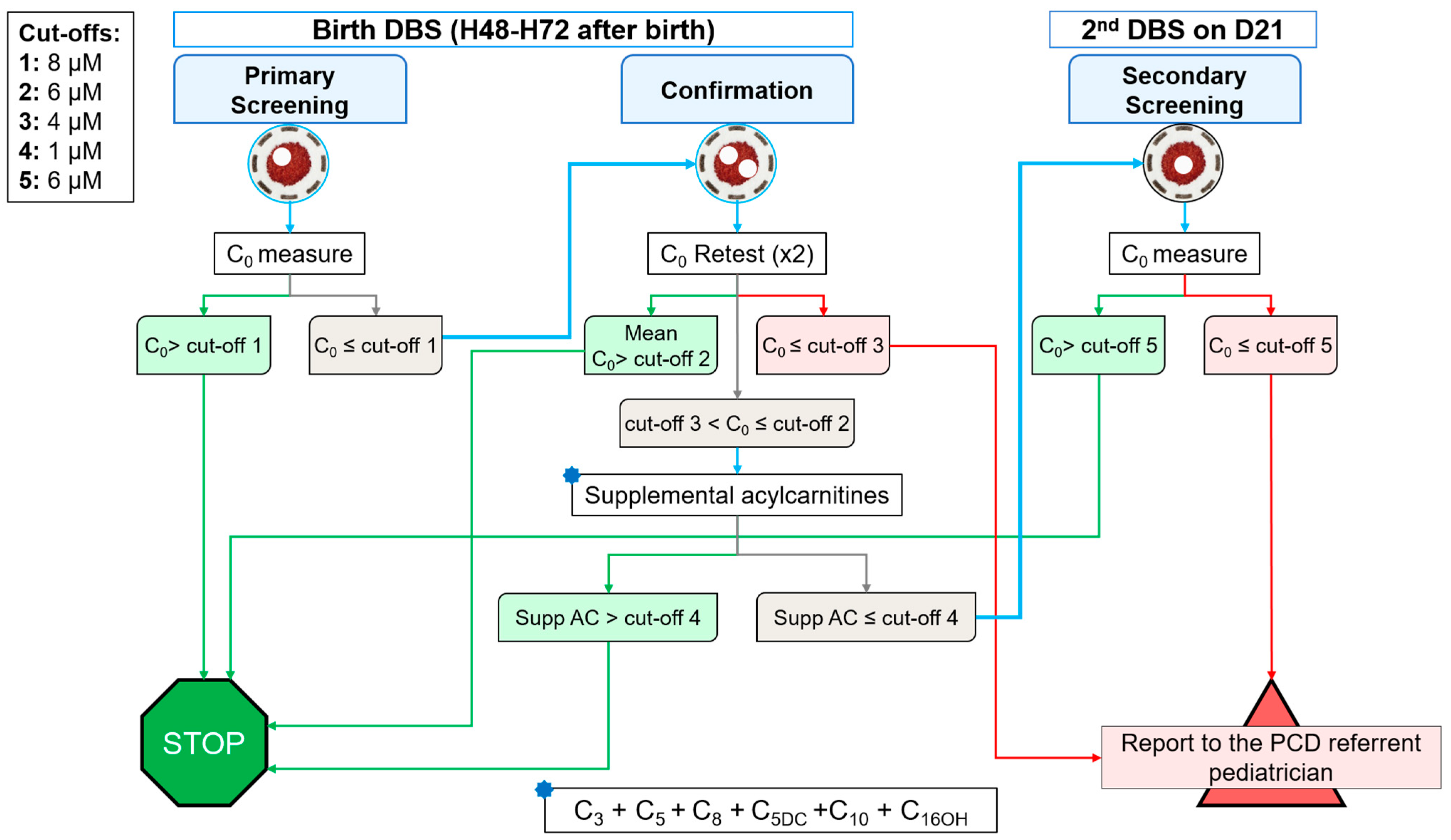
6. Discussion of a Suitable Screening Algorithm
- -
- Using supplemental biomarkers in addition to C0.
- -
- Coupling SLC22A5 molecular testing on initial DBS.
- -
- Performing a retest on a second DBS sample to prevent the impact of the maternal status.
- -
- A C0 higher (>) than cut-off 1 (8 µmol·L−1) means a negative screening, and the patient is ruled-out.
- -
- If C0 is lower or equal (≤) to cut-off 1, a duplicate retest on the birth DBS is performed.
- -
- If mean C0 of this retest is >cut-off 2 (6 µmol·L−1), the patient is ruled-out.
- -
- If mean C0 is ≤cut-off 3 (4 µmol·L−1), the screening is positive and the patient is reported to the PCD referent pediatrician.
- -
- If C0 is between cut-offs 2 and 3, the sum of supplemental acylcarnitines is calculated.
- -
- If supplemental acylcarnitines are >cut-off 4 (1 µmol·L−1), the patient is ruled-out.
- -
- If supplemental acylcarnitines are ≤cut-off 4, another DBS is sampled at day 21 of life.
- -
- If C0 on the 2nd DBS sample is >cut-off 5 (6 µmol·L−1), the patient is ruled-out.
- -
- If C0 is ≤cut-off 5, the screening is positive and the patient is reported to the PCD referent pediatrician.
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, X.; Prasad, P.D.; Leibach, F.H.; Ganapathy, V. CDNA Sequence, Transport Function, and Genomic Organization of Human OCTN2, a New Member of the Organic Cation Transporter Family. Biochem. Biophys. Res. Commun. 1998, 246, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary Systemic Carnitine Deficiency Is Caused by Mutations in a Gene Encoding Sodium Ion-Dependent Carnitine Transporter. Nat. Genet. 1999, 21, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Juraszek, B.; Nałęcz, K.A. SLC22A5 (OCTN2) Carnitine Transporter—Indispensable for Cell Metabolism, a Jekyll and Hyde of Human Cancer. Molecules 2020, 25, 14. [Google Scholar] [CrossRef]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and Functional Identification of Sodium Ion-Dependent, High Affinity Human Carnitine Transporter OCTN2*. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A. Carnitine Deficiency Disorders in Children. Ann. N. Y. Acad. Sci. 2004, 1033, 42–51. [Google Scholar] [CrossRef]
- Longo, N. Primary Carnitine Deficiency and Newborn Screening for Disorders of the Carnitine Cycle. Ann. Nutr. Metab. 2016, 68, 5–9. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Stanley, C.A.; DeLeeuw, S.; Coates, P.M.; Vianey-Liaud, C.; Divry, P.; Bonnefont, J.P.; Saudubray, J.M.; Haymond, M.; Trefz, F.K.; Breningstall, G.N. Chronic Cardiomyopathy and Weakness or Acute Coma in Children with a Defect in Carnitine Uptake. Ann. Neurol. 1991, 30, 709–716. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Disorders of Carnitine Biosynthesis and Transport. Mol. Genet. Metab. 2015, 116, 107–112. [Google Scholar] [CrossRef]
- El-Hattab, A.W. Systemic Primary Carnitine Deficiency. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1993. [Google Scholar]
- Crefcoeur, L.L.; Visser, G.; Ferdinandusse, S.; Wijburg, F.A.; Langeveld, M.; Sjouke, B. Clinical Characteristics of Primary Carnitine Deficiency—A Structured Review Using a Case by Case Approach. J. Inherit. Metab. Dis. 2022, 45, 386–405. [Google Scholar] [CrossRef]
- Rasmussen, J.; Nielsen, O.W.; Janzen, N.; Duno, M.; Køber, L.; Steuerwald, U.; Lund, A.M. Carnitine Levels in 26,462 Individuals from the Nationwide Screening Program for Primary Carnitine Deficiency in the Faroe Islands. J. Inherit. Metab. Dis. 2014, 37, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-Phenotype Correlation in Primary Carnitine Deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, A.; Nozaki, J.; Ohura, T.; Kayo, T.; Wada, Y.; Nezu, J.; Ohashi, R.; Tamai, I.; Shoji, Y.; Takada, G.; et al. Genetic Epidemiology of the Carnitine Transporter OCTN2 Gene in a Japanese Population and Phenotypic Characterization in Japanese Pedigrees with Primary Systemic Carnitine Deficiency. Hum. Mol. Genet. 1999, 8, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; ChB, M. Disorders of the Carnitine Cycle and Detection by Newborn Screening. Ann. Acad. Med. Singap. 2008, 37, 3. [Google Scholar]
- Dobrow, M.J.; Hagens, V.; Chafe, R.; Sullivan, T.; Rabeneck, L. Consolidated Principles for Screening Based on a Systematic Review and Consensus Process. CMAJ Can. Med. Assoc. J. 2018, 190, E422–E429. [Google Scholar] [CrossRef]
- Barns, R.J.; Bowling, F.G.; Brown, G.; Clague, A.E.; Thompson, A. Carnitine in Dried Blood Spots: A Method Suitable for Neonatal Screening. Clin. Chim. Acta 1991, 197, 27–33. [Google Scholar] [CrossRef]
- Wilcken, B.; Wiley, V.; Sim, K.G.; Carpenter, K. Carnitine Transporter Defect Diagnosed by Newborn Screening with Electrospray Tandem Mass Spectrometry. J. Pediatr. 2001, 138, 581–584. [Google Scholar] [CrossRef]
- Bodamer, O.A.; Hoffmann, G.F.; Lindner, M. Expanded Newborn Screening in Europe 2007. J. Inherit. Metab. Dis. 2007, 30, 439–444. [Google Scholar] [CrossRef]
- Sontag, M.K.; Yusuf, C.; Grosse, S.D.; Edelman, S.; Miller, J.I.; McKasson, S.; Kellar-Guenther, Y.; Gaffney, M.; Hinton, C.F.; Cuthbert, C.; et al. Infants with Congenital Disorders Identified Through Newborn Screening—United States, 2015–2017. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1265–1268. [Google Scholar] [CrossRef]
- Koracin, V.; Mlinaric, M.; Baric, I.; Brincat, I.; Djordjevic, M.; Drole Torkar, A.; Fumic, K.; Kocova, M.; Milenkovic, T.; Moldovanu, F.; et al. Current Status of Newborn Screening in Southeastern Europe. Front. Pediatr. 2021, 9, 648939. [Google Scholar] [CrossRef]
- Haute Autorité de Santé: Évaluation a priori de l’extension du dépistage néonatal à une ou plusieurs erreurs innées du métabolisme par spectrométrie de masse en tandem en population générale en France. Available online: https://www.has-sante.fr/jcms/c_2866458/fr/evaluation-a-priori-de-l-extension-du-depistage-neonatal-a-une-ou-plusieurs-erreurs-innees-du-metabolisme-par-spectrometrie-de-masse-en-tandem-volet-2 (accessed on 21 July 2022).
- Care, A.G.D. List of Conditions Screened by Jurisdiction—September 2022. Available online: https://www.health.gov.au/resources/publications/list-of-conditions-screened-by-jurisdiction-september-2022?language=en (accessed on 30 July 2022).
- Wilson, C.; Kerruish, N.J.; Wilcken, B.; Wiltshire, E.; Webster, D. Diagnosis of Disorders of Intermediary Metabolism in New Zealand before and after Expanded Newborn Screening: 2004–2009. N. Z. Med. J. 2012, 125, 42–50. [Google Scholar] [PubMed]
- Wilson, C.; Knoll, D.; de Hora, M.; Kyle, C.; Glamuzina, E.; Webster, D. The Decision to Discontinue Screening for Carnitine Uptake Disorder in New Zealand. J. Inherit. Metab. Dis. 2019, 42, 86–92. [Google Scholar] [CrossRef]
- Therrell, B.L.; Hannon, W.H. National Evaluation of US Newborn Screening System Components. Ment. Retard. Dev. Disabil. Res. Rev. 2006, 12, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R. American College of Medical Genetics Newborn Screening Expert Group Newborn Screening: Toward a Uniform Screening Panel and System—Executive Summary. Pediatrics 2006, 117, S296–S307. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC) Impact of Expanded Newborn Screening--United States, 2006. MMWR Morb. Mortal. Wkly. Rep. 2008, 57, 1012–1015.
- Centers for Disease Control and Prevention (CDC) CDC Grand Rounds: Newborn Screening and Improved Outcomes. MMWR Morb. Mortal. Wkly. Rep. 2012, 61, 390–393.
- Magoulas, P.L.; El-Hattab, A.W. Systemic Primary Carnitine Deficiency: An Overview of Clinical Manifestations, Diagnosis, and Management. Orphanet J. Rare Dis. 2012, 7, 68. [Google Scholar] [CrossRef]
- Ojodu, J.; Singh, S.; Kellar-Guenther, Y.; Yusuf, C.; Jones, E.; Wood, T.; Baker, M.; Sontag, M. NewSTEPs: The Establishment of a National Newborn Screening Technical Assistance Resource Center. Int. J. Neonatal Screen. 2017, 4, 1. [Google Scholar] [CrossRef]
- Hanley, W.B. Newborn Screening in Canada—Are We out of Step? Paediatr. Child Health 2005, 10, 203–207. [Google Scholar] [CrossRef]
- Dyack, S. Expanded Newborn Screening: Lessons Learned from MCAD Deficiency. Paediatr. Child Health 2004, 9, 241–243. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Boutin, M.; Lavoie, P.; Maranda, B. Neonatal Urine Screening Program in the Province of Quebec: Technological Upgrade from Thin Layer Chromatography to Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2021, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, G.J.C. Newborn Screening in Latin America at the Beginning of the 21st Century. J. Inherit. Metab. Dis. 2007, 30, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Cabello, J.F.; Novoa, F.; Huff, H.V.; Colombo, M. Expanded Newborn Screening and Genomic Sequencing in Latin America and the Resulting Social Justice and Ethical Considerations. Int. J. Neonatal Screen. 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, G.J.C. Newborn Screening in Latin America: A Brief Overview of the State of the Art. Am. J. Med. Genet. C Semin. Med. Genet. 2021, 187, 322–328. [Google Scholar] [CrossRef]
- Schulze, A.; Lindner, M.; Kohlmüller, D.; Olgemöller, K.; Mayatepek, E.; Hoffmann, G.F. Expanded Newborn Screening for Inborn Errors of Metabolism by Electrospray Ionization-Tandem Mass Spectrometry: Results, Outcome, and Implications. Pediatrics 2003, 111, 1399–1406. [Google Scholar] [CrossRef]
- Lund, A.M.; Joensen, F.; Hougaard, D.M.; Jensen, L.K.; Christensen, E.; Christensen, M.; Nørgaard-Petersen, B.; Schwartz, M.; Skovby, F. Carnitine Transporter and Holocarboxylase Synthetase Deficiencies in The Faroe Islands. J. Inherit. Metab. Dis. 2007, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Steuerwald, U.; Lund, A.M.; Rasmussen, J.; Janzen, N.; Hougaard, D.M.; Longo, N. Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands. Int. J. Neonatal Screen. 2017, 3, 1. [Google Scholar] [CrossRef]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef]
- Walter, J.H.; Patterson, A.; Till, J.; Besley, G.T.N.; Fleming, G.; Henderson, M.J. Bloodspot Acylcarnitine and Amino Acid Analysis in Cord Blood Samples: Efficacy and Reference Data from a Large Cohort Study. J. Inherit. Metab. Dis. 2009, 32, 95–101. [Google Scholar] [CrossRef]
- Jansen, M.E.; Klein, A.W.; Buitenhuis, E.C.; Rodenburg, W.; Cornel, M.C. Expanded Neonatal Bloodspot Screening Programmes: An Evaluation Framework to Discuss New Conditions With Stakeholders. Front. Pediatr. 2021, 9, 635353. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.C.; Adams, J. Current Status of Newborn Screening Worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Al Hosani, H.; Salah, M.; Osman, H.M.; Farag, H.M.; El Assiouty, L.; Saade, D.; Hertecant, J. Expanding the Comprehensive National Neonatal Screening Programme in the United Arab Emirates from 1995 to 2011. East. Mediterr. Health J. 2014, 20, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Golbahar, J.; Al-Jishi, E.A.; Altayab, D.D.; Carreon, E.; Bakhiet, M.; Alkhayyat, H. Selective Newborn Screening of Inborn Errors of Amino Acids, Organic Acids and Fatty Acids Metabolism in the Kingdom of Bahrain. Mol. Genet. Metab. 2013, 110, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.A.; El-Mougy, F.; Sharaf, S.A.; Mandour, I.; Morgan, M.F.; Selim, L.A.; Hassan, S.A.; Salem, F.; Oraby, A.; Girgis, M.Y.; et al. Inborn Errors of Metabolism Detectable by Tandem Mass Spectrometry in Egypt: The First Newborn Screening Pilot Study. J. Med. Screen. 2016, 23, 124–129. [Google Scholar] [CrossRef]
- Mohamed, S.; Elsheikh, W.; Al-Aqeel, A.I.; Alhashem, A.M.; Alodaib, A.; Alahaideb, L.; Almashary, M.; Alharbi, F.; AlMalawi, H.; Ammari, A.; et al. Incidence of Newborn Screening Disorders among 56632 Infants in Central Saudi Arabia: A 6-Year Study. Saudi Med. J. 2020, 41, 703–708. [Google Scholar] [CrossRef]
- Lindner, M.; Abdoh, G.; Fang-Hoffmann, J.; Shabeck, N.; Al Sayrafi, M.; Al Janahi, M.; Ho, S.; Abdelrahman, M.O.; Ben-Omran, T.; Bener, A.; et al. Implementation of Extended Neonatal Screening and a Metabolic Unit in the State of Qatar: Developing and Optimizing Strategies in Cooperation with the Neonatal Screening Center in Heidelberg. J. Inherit. Metab. Dis. 2007, 30, 522–529. [Google Scholar] [CrossRef]
- Ramaswamy, M.; Anthony Skrinska, V.; Fayez Mitri, R.; Abdoh, G. Diagnosis of Carnitine Deficiency in Extremely Preterm Neonates Related to Parenteral Nutrition: Two Step Newborn Screening Approach. Int. J. Neonatal Screen. 2019, 5, 29. [Google Scholar] [CrossRef]
- Therrell, B.L.; Lloyd-Puryear, M.A.; Ohene-Frempong, K.; Ware, R.E.; Padilla, C.D.; Ambrose, E.E.; Barkat, A.; Ghazal, H.; Kiyaga, C.; Mvalo, T.; et al. Empowering Newborn Screening Programs in African Countries through Establishment of an International Collaborative Effort. J. Community Genet. 2020, 11, 253–268. [Google Scholar] [CrossRef]
- Lamhonwah, A.M.; Tein, I. Carnitine Uptake Defect: Frameshift Mutations in the Human Plasmalemmal Carnitine Transporter Gene. Biochem. Biophys. Res. Commun. 1998, 252, 396–401. [Google Scholar] [CrossRef]
- Tang, N.L.S.; Hwu, W.L.; Chan, R.T.; Law, L.K.; Fung, L.M.; Zhang, W.M. A Founder Mutation (R254X) of SLC22A5 (OCTN2) in Chinese Primary Carnitine Deficiency Patients. Hum. Mutat. 2002, 20, 232. [Google Scholar] [CrossRef]
- Tang, N.; Hui, J. 20 Years After Discovery of the Causative Gene of Primary Carnitine Deficiency, How Much More Have We Known About the Disease? HK J Paediatr New Ser. 2020, 25, 23–29. [Google Scholar]
- Liammongkolkul, S.; Sanomcham, K.; Vatanavicharn, N.; Sathienkijkanchai, A.; Ranieri, E.; Wasant, P. AB133. Expanded Newborn Screening Program in Thailand. Ann. Transl. Med. 2017, 5, AB133. [Google Scholar] [CrossRef]
- Padilla, C.D.; Therrell, B.L.; Alcausin, M.M.L.B.; Chiong, M.A.D.; Abacan, M.A.R.; Reyes, M.E.L.; Jomento, C.M.; Dizon-Escoreal, M.T.T.; Canlas, M.A.E.; Abadingo, M.E.; et al. Successful Implementation of Expanded Newborn Screening in the Philippines Using Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2022, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Mookken, T. Universal Implementation of Newborn Screening in India. Int. J. Neonatal Screen. 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.-Y.; Chen, N.-I.; Chen, P.-W.; Chiang, S.-C.; Hwu, W.-L.; Lee, N.-C.; Chien, Y.-H. Newborn Screening for Citrin Deficiency and Carnitine Uptake Defect Using Second-Tier Molecular Tests. BMC Med. Genet. 2013, 14, 24. [Google Scholar] [CrossRef]
- Huang, X.; Wu, D.; Zhu, L.; Wang, W.; Yang, R.; Yang, J.; He, Q.; Zhu, B.; You, Y.; Xiao, R.; et al. Application of a Next-Generation Sequencing (NGS) Panel in Newborn Screening Efficiently Identifies Inborn Disorders of Neonates. Orphanet J. Rare Dis. 2022, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, W.; Huang, C.; Lin, C.; Lin, W.; Peng, W.; Fu, Q.; Chen, D. Increased Detection of Primary Carnitine Deficiency through Second-Tier Newborn Genetic Screening. Orphanet J. Rare Dis. 2021, 16, 149. [Google Scholar] [CrossRef]
- Volgina, S.Y.; Sokolov, A.A. An Analysis of Medical Care Services for Children With Rare Diseases in the Russian Federation. Front. Pharmacol. 2021, 12, 754073. [Google Scholar] [CrossRef]
- Frazier, D.M.; Millington, D.S.; McCandless, S.E.; Koeberl, D.D.; Weavil, S.D.; Chaing, S.H.; Muenzer, J. The Tandem Mass Spectrometry Newborn Screening Experience in North Carolina: 1997–2005. J. Inherit. Metab. Dis. 2006, 29, 76–85. [Google Scholar] [CrossRef]
- Therrell, B.L.; Lloyd-Puryear, M.A.; Camp, K.M.; Mann, M.Y. Inborn Errors of Metabolism Identified via Newborn Screening: Ten-Year Incidence Data and Costs of Nutritional Interventions for Research Agenda Planning. Mol. Genet. Metab. 2014, 113, 14–26. [Google Scholar] [CrossRef]
- Gallant, N.M.; Leydiker, K.; Wilnai, Y.; Lee, C.; Lorey, F.; Feuchtbaum, L.; Tang, H.; Carter, J.; Enns, G.M.; Packman, S.; et al. Biochemical Characteristics of Newborns with Carnitine Transporter Defect Identified by Newborn Screening in California. Mol. Genet. Metab. 2017, 122, 76–84. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Malvagia, S.; Casetta, B.; Pasquini, E.; Donati, M.A.; Zammarchi, E. Progress in Expanded Newborn Screening for Metabolic Conditions by LC-MS/MS in Tuscany: Update on Methods to Reduce False Tests. J. Inherit. Metab. Dis. 2008, 31, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, L.; Rocha, H.; Sousa, C.; Marcão, A.; Fonseca, H.; Bogas, M.; Osório, R.V. Four Years of Expanded Newborn Screening in Portugal with Tandem Mass Spectrometry. J. Inherit. Metab. Dis. 2010, 33, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.M.; Hougaard, D.M.; Simonsen, H.; Andresen, B.S.; Christensen, M.; Dunø, M.; Skogstrand, K.; Olsen, R.K.J.; Jensen, U.G.; Cohen, A.; et al. Biochemical Screening of 504,049 Newborns in Denmark, the Faroe Islands and Greenland—Experience and Development of a Routine Program for Expanded Newborn Screening. Mol. Genet. Metab. 2012, 107, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Kasper, D.C.; Ratschmann, R.; Metz, T.F.; Mechtler, T.P.; Möslinger, D.; Konstantopoulou, V.; Item, C.B.; Pollak, A.; Herkner, K.R. The National Austrian Newborn Screening Program–Eight Years Experience with Mass Spectrometry. Past, Present, and Future Goals. Wien. Klin. Wochenschr. 2010, 122, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Loukas, Y.L.; Soumelas, G.-S.; Dotsikas, Y.; Georgiou, V.; Molou, E.; Thodi, G.; Boutsini, M.; Biti, S.; Papadopoulos, K. Expanded Newborn Screening in Greece: 30 Months of Experience. J. Inherit. Metab. Dis. 2010, 33, 341–348. [Google Scholar] [CrossRef]
- Lindner, M.; Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Schwab, K.O.; Tacke, U.; Trefz, F.K.; Mengel, E.; Wendel, U.; Leichsenring, M.; et al. Efficacy and Outcome of Expanded Newborn Screening for Metabolic Diseases-Report of 10 Years from South-West Germany *. Orphanet J. Rare Dis. 2011, 6, 44. [Google Scholar] [CrossRef]
- Couce, M.L.; Castiñeiras, D.E.; Bóveda, M.D.; Baña, A.; Cocho, J.A.; Iglesias, A.J.; Colón, C.; Alonso-Fernández, J.R.; Fraga, J.M. Evaluation and Long-Term Follow-up of Infants with Inborn Errors of Metabolism Identified in an Expanded Screening Programme. Mol. Genet. Metab. 2011, 104, 470–475. [Google Scholar] [CrossRef]
- Smon, A.; Repic Lampret, B.; Groselj, U.; Zerjav Tansek, M.; Kovac, J.; Perko, D.; Bertok, S.; Battelino, T.; Trebusak Podkrajsek, K. Next Generation Sequencing as a Follow-up Test in an Expanded Newborn Screening Programme. Clin. Biochem. 2018, 52, 48–55. [Google Scholar] [CrossRef]
- Tangeraas, T.; Sæves, I.; Klingenberg, C.; Jørgensen, J.; Kristensen, E.; Gunnarsdottir, G.; Hansen, E.V.; Strand, J.; Lundman, E.; Ferdinandusse, S.; et al. Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. Int. J. Neonatal Screen. 2020, 6, 51. [Google Scholar] [CrossRef]
- Maguolo, A.; Rodella, G.; Dianin, A.; Nurti, R.; Monge, I.; Rigotti, E.; Cantalupo, G.; Salviati, L.; Tucci, S.; Pellegrini, F.; et al. Diagnosis, Genetic Characterization and Clinical Follow up of Mitochondrial Fatty Acid Oxidation Disorders in the New Era of Expanded Newborn Screening: A Single Centre Experience. Mol. Genet. Metab. Rep. 2020, 24, 100632. [Google Scholar] [CrossRef]
- Lund, A.; Wibrand, F.; Skogstrand, K.; Cohen, A.; Christensen, M.; Jäpelt, R.B.; Dunø, M.; Skovby, F.; Nørgaard-Pedersen, B.; Gregersen, N.; et al. Danish Expanded Newborn Screening Is a Successful Preventive Public Health Programme. Dan. Med. J. 2020, 67, A06190341. [Google Scholar]
- Messina, M.; Meli, C.; Raudino, F.; Pittalá, A.; Arena, A.; Barone, R.; Giuffrida, F.; Iacobacci, R.; Muccilli, V.; Sorge, G.; et al. Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily. Int. J. Neonatal Screen. 2018, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, L.; von Döbeln, U.; Åhlman, H.; Ohlsson, A.; Engvall, M.; Naess, K.; Backman-Johansson, C.; Nordqvist, Y.; Wedell, A.; Zetterström, R.H. Expanded Screening of One Million Swedish Babies with R4S and CLIR for Post-Analytical Evaluation of Data. Int. J. Neonatal Screen. 2020, 6, 42. [Google Scholar] [CrossRef]
- Martín-Rivada, Á.; Palomino Pérez, L.; Ruiz-Sala, P.; Navarrete, R.; Cambra Conejero, A.; Quijada Fraile, P.; Moráis López, A.; Belanger-Quintana, A.; Martín-Hernández, E.; Bellusci, M.; et al. Diagnosis of Inborn Errors of Metabolism within the Expanded Newborn Screening in the Madrid Region. JIMD Rep. 2022, 63, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Conejero, A.C.; Figueras, L.M.; Temprado, A.O.; Soto, P.B.; Martín, Á.; Pérez, L.P.; Villarroya, E.C.; Giner, C.P.; Fraile, P.Q.; Martín-Hernández, E.; et al. Análisis de Casos positivos de cribado neonatal de errores congénitos del metabolismo en la comunidad de madrid. Rev. Esp. Salud. Pública 2020, 94, 15. [Google Scholar]
- Schiergens, K.A.; Weiss, K.J.; Röschinger, W.; Lotz-Havla, A.S.; Schmitt, J.; Dalla Pozza, R.; Ulrich, S.; Odenwald, B.; Kreuder, J.; Maier, E.M. Newborn Screening for Carnitine Transporter Defect in Bavaria and the Long-Term Follow-up of the Identified Newborns and Mothers: Assessing the Benefit and Possible Harm Based on 19 ½ Years of Experience. Mol. Genet. Metab. Rep. 2021, 28, 100776. [Google Scholar] [CrossRef] [PubMed]
- Ruoppolo, M.; Malvagia, S.; Boenzi, S.; Carducci, C.; Dionisi-Vici, C.; Teofoli, F.; Burlina, A.; Angeloni, A.; Aronica, T.; Bordugo, A.; et al. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience. Int. J. Neonatal Screen. 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.-M.; Chien, Y.-H.; Chiang, C.-C.; Ho, H.-C.; Hwu, W.-L.; Kao, S.-M.; Chiang, S.-H.; Kao, C.-H.; Liu, T.-T.; Chiang, H.; et al. Nationwide Survey of Extended Newborn Screening by Tandem Mass Spectrometry in Taiwan. J. Inherit. Metab. Dis. 2010, 33, 295–305. [Google Scholar] [CrossRef]
- Lee, N.-C.; Tang, N.L.-S.; Chien, Y.-H.; Chen, C.-A.; Lin, S.-J.; Chiu, P.-C.; Huang, A.-C.; Hwu, W.-L. Diagnoses of Newborns and Mothers with Carnitine Uptake Defects through Newborn Screening. Mol. Genet. Metab. 2010, 100, 46–50. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Lee, N.-C.; Chao, M.-C.; Chen, L.-C.; Chen, L.-H.; Chien, C.-C.; Ho, H.-C.; Suen, J.-H.; Hwu, W.-L. Fatty Acid Oxidation Disorders in a Chinese Population in Taiwan. In JIMD Reports-Volume 11; Zschocke, J., Gibson, K.M., Brown, G., Morava, E., Peters, V., Eds.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; Volume 11, pp. 165–172. ISBN 978-3-642-37327-5. [Google Scholar]
- Lim, J.S.; Tan, E.S.; John, C.M.; Poh, S.; Yeo, S.J.; Ang, J.S.M.; Adakalaisamy, P.; Rozalli, R.A.; Hart, C.; Tan, E.T.H.; et al. Inborn Error of Metabolism (IEM) Screening in Singapore by Electrospray Ionization-Tandem Mass Spectrometry (ESI/MS/MS): An 8year Journey from Pilot to Current Program. Mol. Genet. Metab. 2014, 113, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Y.-Y.; Jiang, T. Clinical Features and Genotyping of Patients with Primary Carnitine Deficiency Identified by Newborn Screening. J. Pediatr. Endocrinol. Metab. JPEM 2017, 30, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.C.; Law, L.K.; Hui, J.; Lai, C.Y.; Leung, T.Y.; Yuen, Y.P. Expanded Newborn Metabolic Screening Programme in Hong Kong: A Three-Year Journey. Hong Kong Med. J. 2017, 23, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhang, Y.; Hong, F.; Yang, J.; Tong, F.; Mao, H.; Huang, X.; Zhou, X.; Yang, R.; Zhao, Z.; et al. Screening for fatty acid oxidation disorders of newborns in Zhejiang province:prevalence, outcome and follow-up. Zhejiang Xue Xue Bao Yi Xue Ban 2017, 46, 248–255. [Google Scholar]
- Guo, K.; Zhou, X.; Chen, X.; Wu, Y.; Liu, C.; Kong, Q. Expanded Newborn Screening for Inborn Errors of Metabolism and Genetic Characteristics in a Chinese Population. Front. Genet. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Hasegawa, Y.; Yamada, K.; Kobayashi, H.; Purevsuren, J.; Yang, Y.; Dung, V.C.; Khanh, N.N.; Verma, I.C.; Bijarnia-Mahay, S.; et al. Diversity in the Incidence and Spectrum of Organic Acidemias, Fatty Acid Oxidation Disorders, and Amino Acid Disorders in Asian Countries: Selective Screening vs. Expanded Newborn Screening. Mol. Genet. Metab. Rep. 2018, 16, 5–10. [Google Scholar] [CrossRef]
- Kang, E.; Kim, Y.-M.; Kang, M.; Heo, S.-H.; Kim, G.-H.; Choi, I.-H.; Choi, J.-H.; Yoo, H.-W.; Lee, B.H. Clinical and Genetic Characteristics of Patients with Fatty Acid Oxidation Disorders Identified by Newborn Screening. BMC Pediatr. 2018, 18, 103. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Li, H.; Huang, T.; Zhang, Y.; Wang, C.; Gu, M. Biochemical, Molecular, and Clinical Characterization of Patients With Primary Carnitine Deficiency via Large-Scale Newborn Screening in Xuzhou Area. Front. Pediatr. 2019, 7, 50. [Google Scholar] [CrossRef]
- Wang, T.; Ma, J.; Zhang, Q.; Gao, A.; Wang, Q.; Li, H.; Xiang, J.; Wang, B. Expanded Newborn Screening for Inborn Errors of Metabolism by Tandem Mass Spectrometry in Suzhou, China: Disease Spectrum, Prevalence, Genetic Characteristics in a Chinese Population. Front. Genet. 2019, 10, 1052. [Google Scholar] [CrossRef]
- Yang, N.; Gong, L.-F.; Zhao, J.-Q.; Yang, H.-H.; Ma, Z.-J.; Liu, W.; Wan, Z.-H.; Kong, Y.-Y. Inborn Errors of Metabolism Detectable by Tandem Mass Spectrometry in Beijing. J. Pediatr. Endocrinol. Metab. JPEM 2020, 33, 639–645. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, H.; Zhou, D.; Hu, Z.; Zhang, C.; Hu, L.; Zhang, Y.; Zhu, L.; Lu, B.; Zhang, T.; et al. Screening 3.4 Million Newborns for Primary Carnitine Deficiency in Zhejiang Province, China. Clin. Chim. Acta Int. J. Clin. Chem. 2020, 507, 199–204. [Google Scholar] [CrossRef]
- Wang, S.; Leng, J.; Diao, C.; Wang, Y.; Zheng, R. Genetic Characteristics and Follow-up of Patients with Fatty Acid β-Oxidation Disorders through Expanded Newborn Screening in a Northern Chinese Population. J. Pediatr. Endocrinol. Metab. JPEM 2020, 33, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.L.; Tang, C.F.; Liu, S.C.; Sheng, H.Y.; Tang, F.; Jiang, X.; Zheng, R.D.; Mei, H.F.; Liu, L. Newborn screening for primary carnitine deficiency and variant spectrum of SLC22A5 gene in Guangzhou. Zhonghua Er Ke Za Zhi 2020, 58, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, Q.; Zeng, Y.; Qiu, X.; Liu, G.; Zhu, W. Gene Spectrum and Clinical Traits of 10 Patients with Primary Carnitine Deficiency. Mol. Genet. Genomic Med. 2021, 9, e1583. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, Q.; Zheng, T.; Zheng, Z.; Lin, W.; Fu, Q. Expanded Newborn Screening for Inherited Metabolic Disorders and Genetic Characteristics in a Southern Chinese Population. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 494, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Wang, K.; Zheng, Z.; Chen, Y.; Fu, C.; Lin, Y.; Chen, D. Newborn Screening for Primary Carnitine Deficiency in Quanzhou, China. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 512, 166–171. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, B.; Chen, Y.; Zheng, Z.; Fu, Q.; Lin, W.; Zhang, W. Biochemical and Genetic Characteristics of Patients with Primary Carnitine Deficiency Identified through Newborn Screening. Orphanet J. Rare Dis. 2021, 16, 503. [Google Scholar] [CrossRef]
- Yang, X.; Li, Q.; Wang, F.; Yan, L.; Zhuang, D.; Qiu, H.; Li, H.; Chen, L. Newborn Screening and Genetic Analysis Identify Six Novel Genetic Variants for Primary Carnitine Deficiency in Ningbo Area, China. Front. Genet. 2021, 12, 686137. [Google Scholar] [CrossRef]
- Tan, J.; Chen, D.; Chang, R.; Pan, L.; Yang, J.; Yuan, D.; Huang, L.; Yan, T.; Ning, H.; Wei, J.; et al. Tandem Mass Spectrometry Screening for Inborn Errors of Metabolism in Newborns and High-Risk Infants in Southern China: Disease Spectrum and Genetic Characteristics in a Chinese Population. Front. Genet. 2021, 12, 631688. [Google Scholar] [CrossRef]
- Yang, C.; Shi, C.; Zhou, C.; Wan, Q.; Zhou, Y.; Chen, X.; Jin, X.; Huang, C.; Xu, P. Screening and Follow-up Results of Fatty Acid Oxidative Metabolism Disorders in 608 818 Newborns in Jining, Shandong Province. Zhejiang Xue Xue Bao Yi Xue Ban 2021, 50, 472–480. [Google Scholar] [CrossRef]
- Tang, C.; Tan, M.; Xie, T.; Tang, F.; Liu, S.; Wei, Q.; Liu, J.; Huang, Y. Screening for Neonatal Inherited Metabolic Disorders by Tandem Mass Spectrometry in Guangzhou. Zhejiang Xue Xue Bao Yi Xue Ban 2021, 50, 463–471. [Google Scholar] [CrossRef]
- Geng, G.; Yang, Q.; Fan, X.; Lin, C.; Wu, L.; Chen, S.; Luo, J. Analysis of metabolic profile and genetic variants for newborns with primary carnitine deficiency from Guangxi. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2021, 38, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, J.; He, L.; Zeng, Y.; Huang, X.; Luo, Y.; Li, Y. Spectrum Analysis of Inherited Metabolic Disorders for Expanded Newborn Screening in a Central Chinese Population. Front. Genet. 2021, 12, 763222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qiang, R.; Song, C.; Ma, X.; Zhang, Y.; Li, F.; Wang, R.; Yu, W.; Feng, M.; Yang, L.; et al. Spectrum Analysis of Inborn Errors of Metabolism for Expanded Newborn Screening in a Northwestern Chinese Population. Sci. Rep. 2021, 11, 2699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Deng, L.; Huang, Y.; Xiao, Y.; Wen, J.; Liu, N.; Zeng, Y.; Zhang, H. Application of the Artificial Intelligence Algorithm Model for Screening of Inborn Errors of Metabolism. Front. Pediatr. 2022, 10, 855943. [Google Scholar] [CrossRef]
- Luo, X.; Sun, Y.; Xu, F.; Guo, J.; Li, L.; Lin, Z.; Ye, J.; Gu, X.; Yu, Y. A Pilot Study of Expanded Newborn Screening for 573 Genes Related to Severe Inherited Disorders in China: Results from 1,127 Newborns. Ann. Transl. Med. 2020, 8, 1058. [Google Scholar] [CrossRef]
- Frigeni, M.; Balakrishnan, B.; Yin, X.; Calderon, F.R.O.; Mao, R.; Pasquali, M.; Longo, N. Functional and Molecular Studies in Primary Carnitine Deficiency. Hum. Mutat. 2017, 38, 1684–1699. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Li, F.-Y.; Shen, J.; Powell, B.R.; Bawle, E.V.; Adams, D.J.; Wahl, E.; Kobori, J.A.; Graham, B.; Scaglia, F.; et al. Maternal Systemic Primary Carnitine Deficiency Uncovered by Newborn Screening: Clinical, Biochemical, and Molecular Aspects. Genet. Med. 2010, 12, 19–24. [Google Scholar] [CrossRef]
- Lin, H.J.; Neidich, J.A.; Salazar, D.; Thomas-Johnson, E.; Ferreira, B.F.; Kwong, A.M.; Lin, A.M.; Jonas, A.J.; Levine, S.; Lorey, F.; et al. Asymptomatic Maternal Combined Homocystinuria and Methylmalonic Aciduria (CblC) Detected through Low Carnitine Levels on Newborn Screening. J. Pediatr. 2009, 155, 924–927. [Google Scholar] [CrossRef]
- Holme, E.; Jodal, U.; Linstedt, S.; Nordin, I. Effects of Pivalic Acid-Containing Prodrugs on Carnitine Homeostasis and on Response to Fasting in Children. Scand. J. Clin. Lab. Investig. 1992, 52, 361–372. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, H.; Bo, R.; Takahashi, T.; Hasegawa, Y.; Nakamura, M.; Ishige, N.; Yamaguchi, S. Elevation of Pivaloylcarnitine by Sivelestat Sodium in Two Children. Mol. Genet. Metab. 2015, 116, 192–194. [Google Scholar] [CrossRef]
- Peng, G.; Tang, Y.; Cowan, T.M.; Zhao, H.; Scharfe, C. Timing of Newborn Blood Collection Alters Metabolic Disease Screening Performance. Front. Pediatr. 2020, 8, 623184. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Yang, R.; Huang, X.; Tian, Y.; Pei, X.; Bohn, M.K.; Zou, L.; Wang, Y.; Li, H.; Wang, T.; et al. Reference Standards for Newborn Screening of Metabolic Disorders by Tandem Mass Spectrometry: A Nationwide Study on Millions of Chinese Neonatal Populations. Front. Mol. Biosci. 2021, 8, 719866. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Tang, Y.; Gandotra, N.; Enns, G.M.; Cowan, T.M.; Zhao, H.; Scharfe, C. Ethnic Variability in Newborn Metabolic Screening Markers Associated with False-positive Outcomes. J. Inherit. Metab. Dis. 2020, 43, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Reference | Country/Region | Period/ Survey Duration | Newborns Screened | First Tier Test C0 Threshold (µmol·L−1) | Secondary Markers | Number of Patients Diagnosed (Incidence) | Number of Maternal PCD Identified | False Positive Tests (PPV%) | |
|---|---|---|---|---|---|---|---|---|---|
| Australia and New-Zealand | |||||||||
| [18] | New South Wales (Australia) | 1998–2000 | 1,490,000 | <10 (<5 *) | 4 (1:372,500) | ND | 1017 (0.4%) | ||
| [24,25] | New Zealand | 2006–2016 | ~600,000 | <5/5 | 2 (1:300,000) | 9 | 73 (2.7%) | ||
| North America | |||||||||
| [62] | North Carolina (USA) | 1997–2005 | 944,078 | <13 | 0 (<1:944,078) | ND | 0 | ||
| [63] | USA | 2001–2011 | 20,908,664 | ND | 147 (1:142,236) | ||||
| [64] | California (USA) | 2005–2012 | 3,608,768 | <12 (derivatized) | 48 screened (1:75,000) 21 confirmed (1:172,000) | 6 | 1030 (4.7%) | ||
| <7 (underivatized) | |||||||||
| [20] | USA | 2015–2017 | 11,750,856 | ND | 138 (1:85,151) | ||||
| Europe | |||||||||
| [65] | Tuscany (Italy) | 2002–2004 | 160,000 | <8 | 1 (1:160,000) | 1 | 1 (50%) | ||
| [42] | England | 2.5 years | 24,983 | <2 (Cord blood) | 0 (<1:24,983) | 2 | 2 | ||
| [38] | Germany | 1998–2001 | 250,000 | <10 | Sum of (C3–C18) < 5 µmol·L−1 | 1 (1:250,000) | ND | 86 (1.2%) | |
| [66] | Portugal | 4 years | 316,243 | <7 | 4 (1:79,060) | 1 | 1 | ||
| [67] | Danmark Faroe Islands Greenland | 2002–2011 | 504,049 | <5.7 | C5 < 0.43 µmol·L−1 AC/Cit < 3.0 | 5 (1:100,809) | 8 | 28 (15%) | |
| [68] | Austria | 2002–2009 | 622,489 | ND | 2 (1:311,245) | ND | ND | ||
| [69] | Greece | 2007–2009 | 45,000 | <6.25 | 0 (<1:45,000) | ND | ND | ||
| [70] | Germany | 1999–2009 | 1,084,195 | <10 | 3 (1:361,398) | ND | ND | ||
| [71] | Galicia (Spain) | 2000–2015 | 210,165 | <9.5 | 1 (1:210,165) | ND | ND | ||
| [72] | Slovenia | 2013–2014 | 10,048 | <7.7 | AC/Cit < 1.9 | 0 (<1:10,048) | ND | ND | |
| [73] | Norway | 2012–2020 | 461,369 | <6 | C3 + C16 > 2 µmol·L−1 | 3 (1:153,790) | 2 | 22 (12%) | |
| [74] | Verona (Italy) | 2014–2019 | 86,320 | ND | 3 (1:28,773) | ND | ND | ||
| [75] | Danmark Faroe Islands Greenland | 2002–2018 | 967,780 | C0 < 5.7 | C5 < 0.43 µmol·L−1 Ac/Cit < 3.0 | 40 (1:24,195) 32 TP + 8 FN | 19 | 114 (21.9%) | |
| [76] | Sicilia | 2011–2017 | 60,408 | ND | 0 (1:60,408) | ND | ND | ||
| [77] | Sweden | 2011–2019 | 1,000,000 | ND | 13 (1:76,923) | 6 | 94 (12%) | ||
| [78,79] | Madrid (Spain) | 2011–2019 | 592,822 | C0 < ND AC/Cit < ND | 12 (1:49,402) | ND | 73 (14%) | ||
| [80] | Bavaria (Germany) | 1999–2018 | 1,816,000 | <9 | 6 (1:302,667) | 12 | 151 (3.8%) | ||
| [81] | Italy | 2017–2020 | 806,770 | ND | Total AC | 10 (1:80,677) | 20 | ND | |
| Asia | |||||||||
| [82] | Taiwan | 2000–2009 | 592,717 | <8 Or <5 | (<2 *) | Recall DBS | 5 confirmed (1:118,543) +2 unconfirmed | 0 | 111 (4.3%) |
| [83] | Taiwan | 2001–2005 | 304,536 | <2.6 <2.86 <10.95 <6.44 <8 | Recall DBS | 4 (1:67,000) ‡ | 6 | 12 (25%) | |
| 2006 | 88,200 | ||||||||
| 2007/1–2007/5 | 31,329 | ||||||||
| 2007/6–2007/12 | 59,785 | ||||||||
| 2008/1–2009/7 | 110,962 | ||||||||
| [84] | Taiwan | 2003–2012 | 790,569 | <6.44 | 22 (1:35,934) | 12 | ND | ||
| [58] | Taiwan | 6 months | 30,237 | <12 (<6.0 *) | 1 (1:30,237) | 0 | 209 (0.48%) | ||
| [85] | Singapore | 2006–2014 | 117,267 | <8 | C2 < 7 µmol·L−1 | 5 (1:35,453) | 5 | 20 (20%) | |
| [86] | Nanjing (China) | 2013–2016 | 62,568 | <10 | 7 (1:8,938) | ND | ND | ||
| [87] | Hong Kong | 2013–2016 | 30,448 | <6.4 | 0 (<1:30448) | 1 | 17 | ||
| [88] | Zhejiang (China) | 2009–2016 | 1,861,262 | ND | 78 (1:23,350) | ND | ND | ||
| [55] | Thailand | 2014–2017 | 99,234 | ND | 5 (1: 372,252) | 6 | ND | ||
| [89] | Jining (China) | 2015 | 48,287 | ND | 5 (1:9,657) | ND | ND | ||
| [90] | Japan | 1997–2015 | 3,360,000 | ND | 17 (1:199,000) | ND | ND | ||
| Taiwan | 2001–2014 | 1,390,000 | 20 (1:70,000) | ||||||
| South Korea | 2000–2015 | 3,440,000 | 10 (1:345,000) | ||||||
| Germany | 2002–2015 | 7,510,000 | 30 (1:250,000) | ||||||
| [91] | Seoul (South Korea) | 2002–2016 | ND | <12 | 1 (ND) | ND | ND | ||
| [92] | Xuzhou (China) | 2015–2017 | 236,368 | <9.63 (<5 *) | 10 (1:23,637) | 6 | 176 (5.4%) | ||
| [93] | Suzhou (China) | 2014–2018 | 401,660 | <9.5 | 15 (1:26,777) | ND | ND | ||
| [94] | Beijing (China) | 2014–2019 | 58,651 | <10 | 1 (1:58,651) | ND | ND | ||
| [95] | Zhejiang (China) | 2009–2019 | 3,410,600 | <14 (derivatized) | 113 (1:30,182) | 63 | ND | ||
| <10.28 (underivatized) | |||||||||
| [96] | Tianjin (China) | 2013–2018 | 220,443 | <13 (derivatized) | 10 (1:22,044) | ND | ND | ||
| [97] | Guangzhou (China) | 2015–2019 | 200,180 | <10 | C0 < 8.5 µmol·L−1 Or C0 [8.5–10] And C3 + C16 < 2 µmol·L−1 | 15 (1:13,345) | 22 | 239 (5.9%) | |
| [98] | Fujian (China) | 2015–2020 | 94,453 | <8.8 | 9 (1:10,495) | 1 | ND | ||
| [60,99,100,101] | Quanzhou (China) | 2014–2021 | 548,247 | <8.5 | 49 (1:11,189) | 6 | 1665 (2.9%) | ||
| [102] | Ningbo (China) | 2014–2018 | 265,524 | <9.5 | 16 (1:16,595) | 3 confirmed + 7 unconfirmed | 1669 (0.96%) | ||
| [103] | Liuzhou (China) | 2012–2020 | 111,986 | <9 | 12 (1:9,332) | 2452 (0.49%) | |||
| [104] | Jining (China) | 2014–2019 | 608,818 | <10 | 16 (1:38,051) | ND | ND | ||
| [105] | Guangzhou (China) | 2015–2020 | 272,117 | <10 | (1) C0 < 8.5 µmol·L−1 Or (2) C0 [8.5–10] And C3 + C16 < 2 µmol·L−1 | 21 (1:12,958) | 30 | (1) 314 (8.7%) (2) 165 (15.3%) | |
| [106] | Guangxi (China) | 2014–2018 | 400,575 | ND | 22 (1:18,208) | 9 | ND | ||
| [107] | Changsha (China) | 2016–2020 | 300,849 | <8.5 | 22 (13,675) | ND | ND | ||
| [108] | Shaanxi (China) | 2014–2019 | 146,152 | <8.5 | 3 (1:48,717) | 2 | ND | ||
| [56] | Philippines | 2005–2011 | 111,127 | ND | 0 (<1:111,127) | ND | ND | ||
| [109] | Shaoyang (Chine) | 2016–2020 | 94,648 | ND | 5 (1:18,930) | ND | 474 (1%) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefèvre, C.R.; Labarthe, F.; Dufour, D.; Moreau, C.; Faoucher, M.; Rollier, P.; Arnoux, J.-B.; Tardieu, M.; Damaj, L.; Bendavid, C.; et al. Newborn Screening of Primary Carnitine Deficiency: An Overview of Worldwide Practices and Pitfalls to Define an Algorithm before Expansion of Newborn Screening in France. Int. J. Neonatal Screen. 2023, 9, 6. https://doi.org/10.3390/ijns9010006
Lefèvre CR, Labarthe F, Dufour D, Moreau C, Faoucher M, Rollier P, Arnoux J-B, Tardieu M, Damaj L, Bendavid C, et al. Newborn Screening of Primary Carnitine Deficiency: An Overview of Worldwide Practices and Pitfalls to Define an Algorithm before Expansion of Newborn Screening in France. International Journal of Neonatal Screening. 2023; 9(1):6. https://doi.org/10.3390/ijns9010006
Chicago/Turabian StyleLefèvre, Charles R., François Labarthe, Diane Dufour, Caroline Moreau, Marie Faoucher, Paul Rollier, Jean-Baptiste Arnoux, Marine Tardieu, Léna Damaj, Claude Bendavid, and et al. 2023. "Newborn Screening of Primary Carnitine Deficiency: An Overview of Worldwide Practices and Pitfalls to Define an Algorithm before Expansion of Newborn Screening in France" International Journal of Neonatal Screening 9, no. 1: 6. https://doi.org/10.3390/ijns9010006