
Low Psychosine in Krabbe Disease with Onset in Late Infancy: A Case Report

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
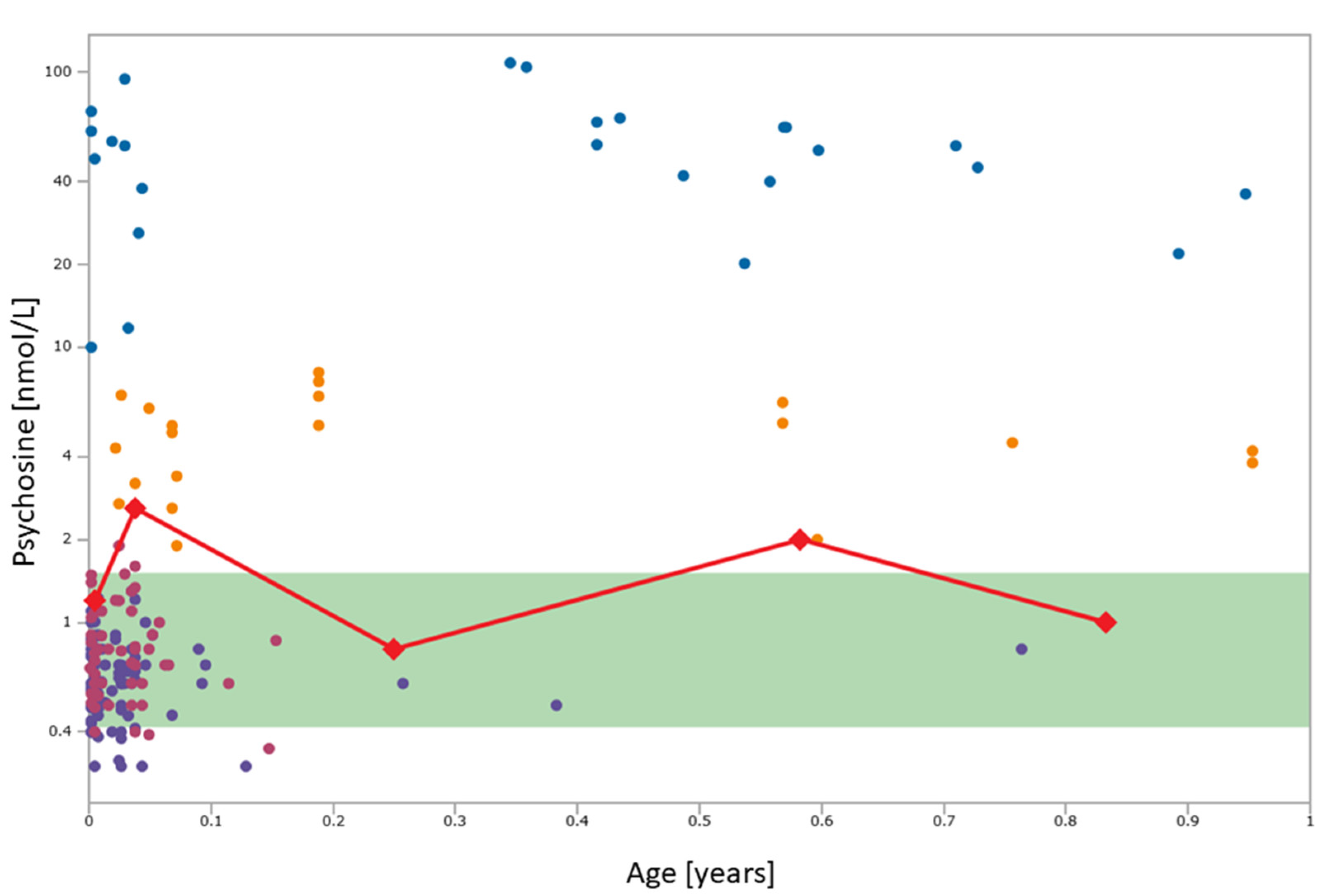
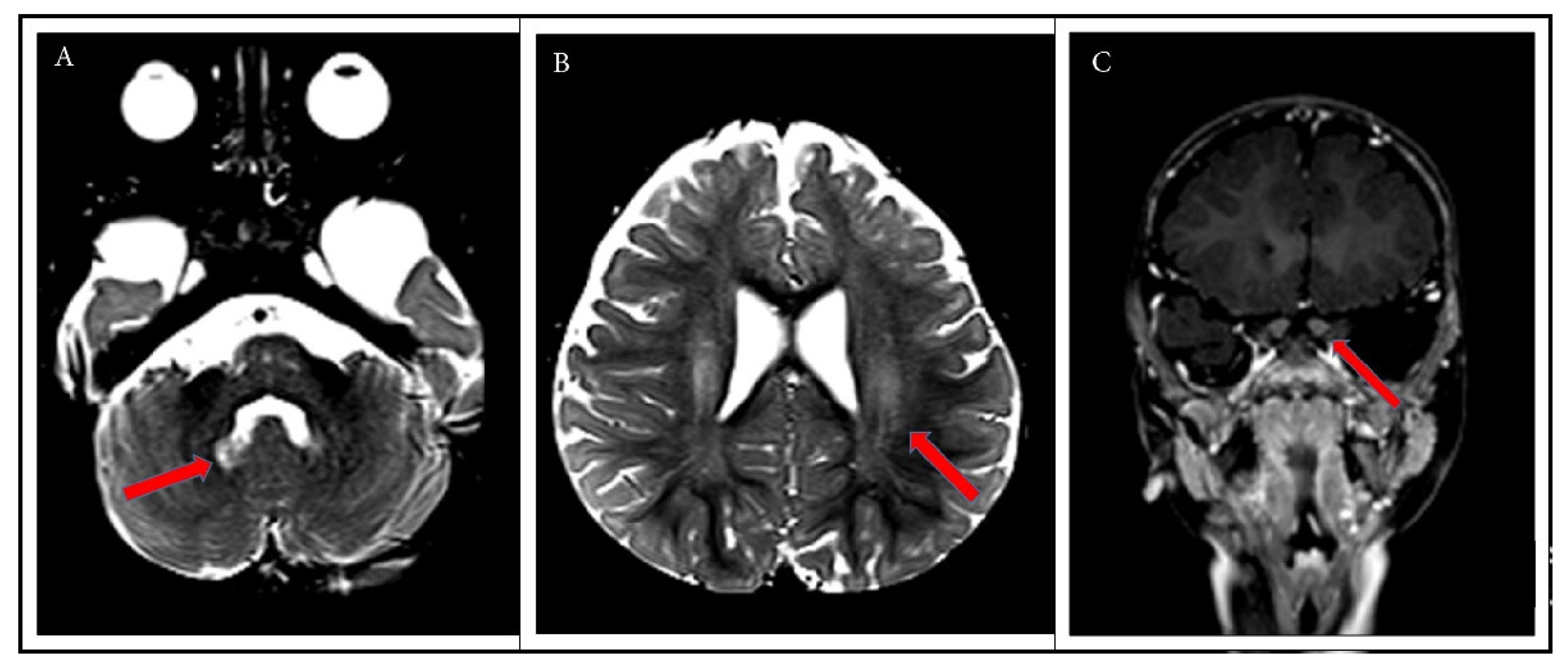
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wenger, D.A.; Rafi, M.A.; Luzi, P. Krabbe Disease: One Hundred Years from the Bedside to the Bench to the Bedside. J. Neurosci. Res. 2016, 94, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Gelinas, J.; Sirrs, S. Phenotypic Variability of Krabbe Disease across the Lifespan. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2014, 41, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Allewelt, H.; Taskindoust, M.; Troy, J.; Page, K.; Wood, S.; Parikh, S.; Prasad, V.K.; Kurtzberg, J. Long-Term Functional Outcomes after Hematopoietic Stem Cell Transplant for Early Infantile Krabbe Disease. Biol. Blood Marrow Transplant. 2018, 24, 2233–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escolar, M.L.; Poe, M.D.; Provenzale, J.M.; Richards, K.C.; Allison, J.; Wood, S.; Wenger, D.A.; Pietryga, D.; Wall, D.; Champagne, M.; et al. Transplantation of Umbilical-Cord Blood in Babies with Infantile Krabbe’s Disease. N. Engl. J. Med. 2005, 352, 2069–2081. [Google Scholar] [CrossRef] [Green Version]
- Wright, M.D.; Poe, M.D.; DeRenzo, A.; Haldal, S.; Escolar, M.L. Developmental Outcomes of Cord Blood Transplantation for Krabbe Disease: A 15-Year Study. Neurology 2017, 89, 1365–1372. [Google Scholar] [CrossRef]
- Tortorelli, S.; Turgeon, C.T.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Rinaldo, P.; Matern, D. Simultaneous Testing for 6 Lysosomal Storage Disorders and X-Adrenoleukodystrophy in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2016, 62, 1248–1254. [Google Scholar] [CrossRef] [Green Version]
- Orsini, J.J.; Kay, D.M.; Saavedra-Matiz, C.A.; Wenger, D.A.; Duffner, P.K.; Erbe, R.W.; Biski, C.; Martin, M.; Krein, L.M.; Nichols, M.; et al. Newborn Screening for Krabbe Disease in New York State: The First Eight Years’ Experience. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 18, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York Pilot Newborn Screening Program for Lysosomal Storage Diseases: Report of the First 65,000 Infants. Genet. Med. J. Am. Coll. Med. Genet. 2019, 21, 631–640. [Google Scholar] [CrossRef]
- Escolar, M.L.; Kiely, B.T.; Shawgo, E.; Hong, X.; Gelb, M.H.; Orsini, J.J.; Matern, D.; Poe, M.D. Psychosine, a Marker of Krabbe Phenotype and Treatment Effect. Mol. Genet. Metab. 2017, 121, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Kim, M.W.; Kim, S.U. Tissue Culture Model of Krabbe’s Disease: Psychosine Cytotoxicity in Rat Oligodendrocyte Culture. Dev. Neurosci. 1997, 19, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Haq, E.; Giri, S.; Singh, I.; Singh, A.K. Molecular Mechanism of Psychosine-Induced Cell Death in Human Oligodendrocyte Cell Line. J. Neurochem. 2003, 86, 1428–1440. [Google Scholar] [CrossRef]
- Li, Y.; Xu, Y.; Benitez, B.A.; Nagree, M.S.; Dearborn, J.T.; Jiang, X.; Guzman, M.A.; Woloszynek, J.C.; Giaramita, A.; Yip, B.K.; et al. Genetic Ablation of Acid Ceramidase in Krabbe Disease Confirms the Psychosine Hypothesis and Identifies a New Therapeutic Target. Proc. Natl. Acad. Sci. USA 2019, 116, 20097–20103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, W.-L.; Pacheco, J.; Zhang, X.K.; Martin, M.M.; Biski, C.K.; Keutzer, J.M.; Wenger, D.A.; Caggana, M.; Orsini, J.J. Determination of Psychosine Concentration in Dried Blood Spots from Newborns That Were Identified via Newborn Screening to Be at Risk for Krabbe Disease. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 419, 73–76. [Google Scholar] [CrossRef]
- Turgeon, C.T.; Orsini, J.J.; Sanders, K.A.; Magera, M.J.; Langan, T.J.; Escolar, M.L.; Duffner, P.; Oglesbee, D.; Gavrilov, D.; Tortorelli, S.; et al. Measurement of Psychosine in Dried Blood Spots—A Possible Improvement to Newborn Screening Programs for Krabbe Disease. J. Inherit. Metab. Dis. 2015, 38, 923–929. [Google Scholar] [CrossRef]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision Newborn Screening for Lysosomal Disorders. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.M.; Matern, D.; Kurtzberg, J.; Wrabetz, L.; Gelb, M.H.; Wenger, D.A.; Ficicioglu, C.; Waldman, A.T.; Burton, B.K.; Hopkins, P.V.; et al. Consensus Guidelines for Newborn Screening, Diagnosis and Treatment of Infantile Krabbe Disease. Orphanet J. Rare Dis. 2018, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenzel, A.J.; Turgeon, C.T.; Nickander, K.K.; White, A.L.; Peck, D.S.; Pino, G.B.; Studinski, A.L.; Prasad, V.K.; Kurtzberg, J.; Escolar, M.L.; et al. The Critical Role of Psychosine in Screening, Diagnosis, and Monitoring of Krabbe Disease. Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 1108–1118. [Google Scholar] [CrossRef]
- Thompson-Stone, R.; Ream, M.A.; Gelb, M.; Matern, D.; Orsini, J.J.; Levy, P.A.; Rubin, J.P.; Wenger, D.A.; Burton, B.K.; Escolar, M.L.; et al. Consensus recommendations for the classification and long-term followup of infants who screen positive for Krabbe Disease. Mol. Genet. Metab. 2021. [Google Scholar] [CrossRef]
- Bascou, N.; DeRenzo, A.; Poe, M.D.; Escolar, M.L. A Prospective Natural History Study of Krabbe Disease in a Patient Cohort with Onset between 6 Months and 3 Years of Life. Orphanet J. Rare Dis. 2018, 13, 46. [Google Scholar] [CrossRef]
- Wenger, D.A.; Rafi, M.A.; Luzi, P. Molecular Genetics of Krabbe Disease (Globoid Cell Leukodystrophy): Diagnostic and Clinical Implications. Hum. Mutat. 1997, 10, 268–279. [Google Scholar] [CrossRef]
- Tappino, B.; Biancheri, R.; Mort, M.; Regis, S.; Corsolini, F.; Rossi, A.; Stroppiano, M.; Lualdi, S.; Fiumara, A.; Bembi, B.; et al. Identification and Characterization of 15 Novel GALC Gene Mutations Causing Krabbe Disease. Hum. Mutat. 2010, 31, E1894–E1914. [Google Scholar] [CrossRef] [Green Version]
- Duffner, P.K.; Barczykowski, A.; Jalal, K.; Yan, L.; Kay, D.M.; Carter, R.L. Early Infantile Krabbe Disease: Results of the World-Wide Krabbe Registry. Pediatr. Neurol. 2011, 45, 141–148. [Google Scholar] [CrossRef]
- Szymańska, K.; Ługowska, A.; Laure-Kamionowska, M.; Bekiesińska-Figatowska, M.; Gieruszczak-Białek, D.; Musielak, M.; Eichler, S.; Giese, A.-K.; Rolfs, A. Diagnostic Difficulties in Krabbe Disease: A Report of Two Cases and Review of Literature. Folia Neuropathol. 2012, 50, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran-Quintero, M.L.; Bascou, N.A.; Poe, M.D.; Wenger, D.A.; Saavedra-Matiz, C.A.; Nichols, M.J.; Escolar, M.L. Early Progression of Krabbe Disease in Patients with Symptom Onset between 0 and 5 Months. Orphanet J. Rare Dis. 2019, 14, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, J.E.; Graham, S.C.; Kim, N.N.; Stein, P.E.; McNair, R.; Cachón-González, M.B.; Cox, T.M.; Read, R.J. Insights into Krabbe Disease from Structures of Galactocerebrosidase. Proc. Natl. Acad. Sci. USA 2011, 108, 15169–15173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saavedra-Matiz, C.A.; Luzi, P.; Nichols, M.; Orsini, J.J.; Caggana, M.; Wenger, D.A. Expression of Individual Mutations and Haplotypes in the Galactocerebrosidase Gene Identified by the Newborn Screening Program in New York State and in Confirmed Cases of Krabbe’s Disease. J. Neurosci. Res. 2016, 94, 1076–1083. [Google Scholar] [CrossRef]
- Spratley, S.J.; Hill, C.H.; Viuff, A.H.; Edgar, J.R.; Skjødt, K.; Deane, J.E. Molecular Mechanisms of Disease Pathogenesis Differ in Krabbe Disease Variants. Traffic Cph. Den. 2016, 17, 908–922. [Google Scholar] [CrossRef]
- Shin, D.; Feltri, M.L.; Wrabetz, L. Altered Trafficking and Processing of GALC Mutants Correlates with Globoid Cell Leukodystrophy Severity. J. Neurosci. J. Soc. Neurosci. 2016, 36, 1858–1870. [Google Scholar] [CrossRef] [Green Version]
- Herbst, Z.; Turgeon, C.T.; Biski, C.; Khaledi, H.; Shoemaker, N.B.; DeArmond, P.D.; Smith, S.; Orsini, J.; Matern, D.; Gelb, M.H. Achieving Congruence among Reference Laboratories for Absolute Abundance Measurement of Analytes for Rare Diseases: Psychosine for Diagnosis and Prognosis of Krabbe Disease. Int. J. Neonatal Screen. 2020, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffner, P.K.; Caggana, M.; Orsini, J.J.; Wenger, D.A.; Patterson, M.C.; Crosley, C.J.; Kurtzberg, J.; Arnold, G.L.; Escolar, M.L.; Adams, D.J.; et al. Newborn Screening for Krabbe Disease: The New York State Model. Pediatr. Neurol. 2009, 40, 245–252, discussion 253–255. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corre, C.S.; Matern, D.; Pellegrino, J.E.; Saavedra-Matiz, C.A.; Orsini, J.J.; Thompson-Stone, R. Low Psychosine in Krabbe Disease with Onset in Late Infancy: A Case Report. Int. J. Neonatal Screen. 2021, 7, 28. https://doi.org/10.3390/ijns7020028
Corre CS, Matern D, Pellegrino JE, Saavedra-Matiz CA, Orsini JJ, Thompson-Stone R. Low Psychosine in Krabbe Disease with Onset in Late Infancy: A Case Report. International Journal of Neonatal Screening. 2021; 7(2):28. https://doi.org/10.3390/ijns7020028
Chicago/Turabian StyleCorre, Camille S., Dietrich Matern, Joan E. Pellegrino, Carlos A. Saavedra-Matiz, Joseph J. Orsini, and Robert Thompson-Stone. 2021. "Low Psychosine in Krabbe Disease with Onset in Late Infancy: A Case Report" International Journal of Neonatal Screening 7, no. 2: 28. https://doi.org/10.3390/ijns7020028