1. Introduction
Since their market launch in 1991 [
1], lithium-ion batteries (LIBs) have developed into an everyday energy storage device, powering almost all mobile electronic devices [
2]. Graphite has been the anode material of choice, despite a comparably low gravimetric capacity of 372 mAh
. Even as early as in the 1960s, about 30 years before the market launch of commercial LIBs, silicon (Si) has been known to be able to store 3579 mAh
or even up to 4212 mAh
via electrochemical alloying to Li
15Si
4 or Li
22Si
5, respectively. This is more than ten times the capacity of graphite [
3]. At the same time, silicon has a comparable or even lower mass density than graphite [
3], rendering it a potentially interesting anode material. However, the volume of silicon changes drastically during (de)lithiation, introducing several problems, e.g., mechanical cracking and disintegration, loss of electric contact, or breakdown of the solid-electrolyte interphase (SEI) [
3].
Silicon’s high specific capacity is utilized in composite anodes consisting of both graphite and silicon. By adding small amounts of silicon to conventional graphite anodes, their capacity can be increased. Such composite anodes have been investigated and commercialized already [
4,
5,
6,
7,
8,
9]. Pure-silicon and Si-dominant anodes, on the other hand, usually are mechanically unstable due to the large volume changes of up to 300% upon (de)lithiation [
10]. Ko et al. [
10] summarized several approaches to manage this and to enable pure-silicon and silicon-dominant anodes as a successor to graphite anodes. The approaches include size-controlled materials, patterned thin films, shape-preserving shell designs, porous structures, and graphene composites. The silicon anode used in this work belongs to the category of porous structures. In the work of Ko et al. [
10], most of the porous structures were synthesized via electrochemical etching of a silicon wafer resulting in nanometer-sized pores. The silicon in this work, however, consists of micrometer-sized silicon particles prepared via jet milling of photovoltaic grade silicon lumps [
11]. Obrovac and Krause [
12] proposed partial lithiation as another approach to increase cycle life and maintain electrode integrity while still providing higher gravimetric capacities than graphite at full lithiation. This concept of partial lithiation also holds true for the electrode design used in this work, as shown by Jantke et al. [
11]. Haufe et al. [
13] investigated the failure mechanism of this electrode design with partially lithiated Si. They found that the main failure mechanism is the continuous formation of SEI. Another major contribution is found in particle decoupling when operating at low discharge voltages.
With an appropriate model, an estimation about (im)practical cell designs without the need for time- and cost-intensive experiments is possible. Doyle et al. [
14] proposed the dual insertion model, also known as the pseudo-2D (p2D) model or Newman model, which is capable of describing the electrochemical behavior of a rocking-chair cell. Since its publication in 1993, the Newman model and its variations have become a famous and widely used approach for physicochemical modeling of LIBs. For example, it has been used to describe relaxation phenomena [
15], short-circuit events [
16], or to aid cell design [
17]. Additionally, the original publication by Doyle et al. [
14] has gained increased research interest over the years, receiving 20% of all citations in the last two years. However, the model first and foremost considers insertion reactions. Hence, care should be taken when using the p2D model for alloying materials, because surface processes (electrode kinetics) as well as transport processes inside the particles (solid phase diffusion) might differ from the implemented processes in the standard p2D model.
Table 1 lists a selection of different simulation models all related to silicon. The models found in the literature cover a wide range of different application scenarios and a variety of different model classes, such as finite element method model, density functional theory models, or impedance-based models, can be found.
When it comes to electrochemical modeling of silicon electrodes, the literature provides different approaches, most of which rely on the Butler–Volmer equation as a reaction kinetic and Fickian solid phase diffusion. Jiang et al. [
18], for example, presented a model depicting the different lithiation stages of silicon. At the same time, different activation energies allow for the representation of crystallization effects and different particle sizes can be accounted for by varying equilibrium potentials. Lory et al. [
19] presented an adapted p2D model for a Si-graphite composite anode. The model considers three different active material phases; graphite, a carbon matrix, and nano-silicon embedded in the carbon matrix. Thus, the influence of silicon on the cell behavior can be modeled and investigated. Swamy and Chiang [
20] published an impedance-based equivalent circuit model to compare the charge transfer overpotentials of pure silicon particles with those of pure graphite particles with respect to the C-rate and the individual particle radius. According to their results, overpotentials may become rate limiting in dependence on the exchange current density.
Regarding the solid phase diffusion, Obrovac and Krause [
12] proposed that a partially lithiated silicon particle would show a core–shell structure with the lithiated amorphous phase as the outer layer and a crystalline core. Such a core–shell structure was also detected by McDowell et al. [
31] during a lithation process of a single particle using in-situ transmission electron microscopy measurements. To properly capture the impacts of this core–shell structure in a model, Wang et al. [
21] used a two-dimensional single particle diffusion model which also considers a reaction front at the phase boundary. Doing so, they could replicate the experimental results. Such a phase boundary is not readily available for the standard p2D model. However, Bordes et al. [
32] investigated polycrystalline silicon particles via transmission electron microscopy and found that lithiation preferentially occurs along defects. The particle then consists of several smaller crystalline cores separated by amorphous “veins”. Such a structure is similar to the porous structure of the electrode where the liquid diffusion is described by an effective diffusion coefficient [
14]. In this analogy, the pore space filled with electrolyte is equivalent to the amorphous Si phase, and the solid-phase structure of the electrode is equivalent to the crystalline cores which hinder the overall transport. Thus, from a modeling perspective, such a structure can be described by an effective diffusion coefficient without the need for a moving phase boundary, which can be accomplished with the standard p2D model.
Other models in the literature focus not on electrochemistry but on mechanics. This work does not focus on mechanics and the interested reader is referred to the literature given in
Table 1, especially references [
22,
24,
25,
26,
29].
The aim of this work is to provide a parametrized and validated p2D model for a silicon-dominant electrode, since to the best knowledge of the authors, the literature lacks such a model. The anode coating in this work contains graphite, but it is largely electrochemically inactive and mainly serves as a conductive additive [
11]. Thus, the electrode is considered silicon-dominant. The loading of 3.61 mAh cm
−2 is application-oriented. Furthermore, the silicon particle diameter is in the micrometer range [
11], which has hardly been studied in literature, especially not simulatively. Finally, it is important to mention that the silicon is partially lithiated to only approximately 30% (i.e., approximately 1200 mAh
). This benefits the mechanical integrity and thus the cycling stability of the electrode [
11].
For a systematic presentation of its contents, this work is structured as follows. First, the properties of the electrodes used are presented. Second, a literature review for material parameters that could not be determined otherwise is discussed. Third, measurements conducted at coin cells and T-cells are presented and discussed. The equilibrium potential measurements of the silicon electrode are of particular interest here. Fourth, the reconstruction of the full-cell quasi open-circuit potential (qOCP) from measured half-cell qOCP curves is discussed, as the curves are a required model input. In a fifth step, a parameter fitting is performed and validated against the full-cell potential at C-rates up to for charging and discharging. Finally, the model accuracy is evaluated and recommendations for further work are drawn.
2. Experimental Procedure
The experimental methods in this work are split into three parts: Electrode manufacturing, cell assembly, and operational procedures. This work includes all aspects of these three steps necessary to perform parametrization and simulation of a silicon-dominant anode. A more detailed description to each of these steps can be found in the
Supplementary Materials.
Electrode manufacturing was performed by the
iwb at the Technical University of Munich. Silicon material was provided by the manufacturer. The preparation of the coating slurry as well as coating and drying of electrode sheets was performed according to the recipe provided by Jantke et al. [
11]. The anode coating comprises of
micrometer Si particles,
graphite, and
binders and additives. Note that the graphite is electrochemically inactive and only serves as a conductive agent [
11], hence the naming of a silicon-dominant anode. The cathode active material is a commercially available nickel-rich nickel-cobalt-aluminum-oxide (NCA) with a composition of LiNi
0.8Co
0.15Al
0.05O
1.985(BO
3)
0.01. As already mentioned, a more detailed description of further electrode properties related to the manufacturing step can be found in
Section S1 along with
Tables S1 and S2 of the Supplementary Materials.
The assembled cells are circular coin cells (CR2032) for characterization as well as Swagelok
® (Solon, OH, USA) T-cells for validation. Coin cells were assembled as half cells vs. lithium (Li) metal as well as full cells. The data retrieved from the coin cells was used for characterization, especially to retrieve the half-cell equilibrium potentials using an electrode balancing process, which is discussed below. Additionally, Swagelok
® T-cells using a three-electrode configuration were assembled as full cells with a Li metal reference electrode. The T-cells were used for validation at higher C-rates. Detailed information on the cell assembly process as well as the separator and electrolyte used are given in
Section S2 of the Supplementary Materials.
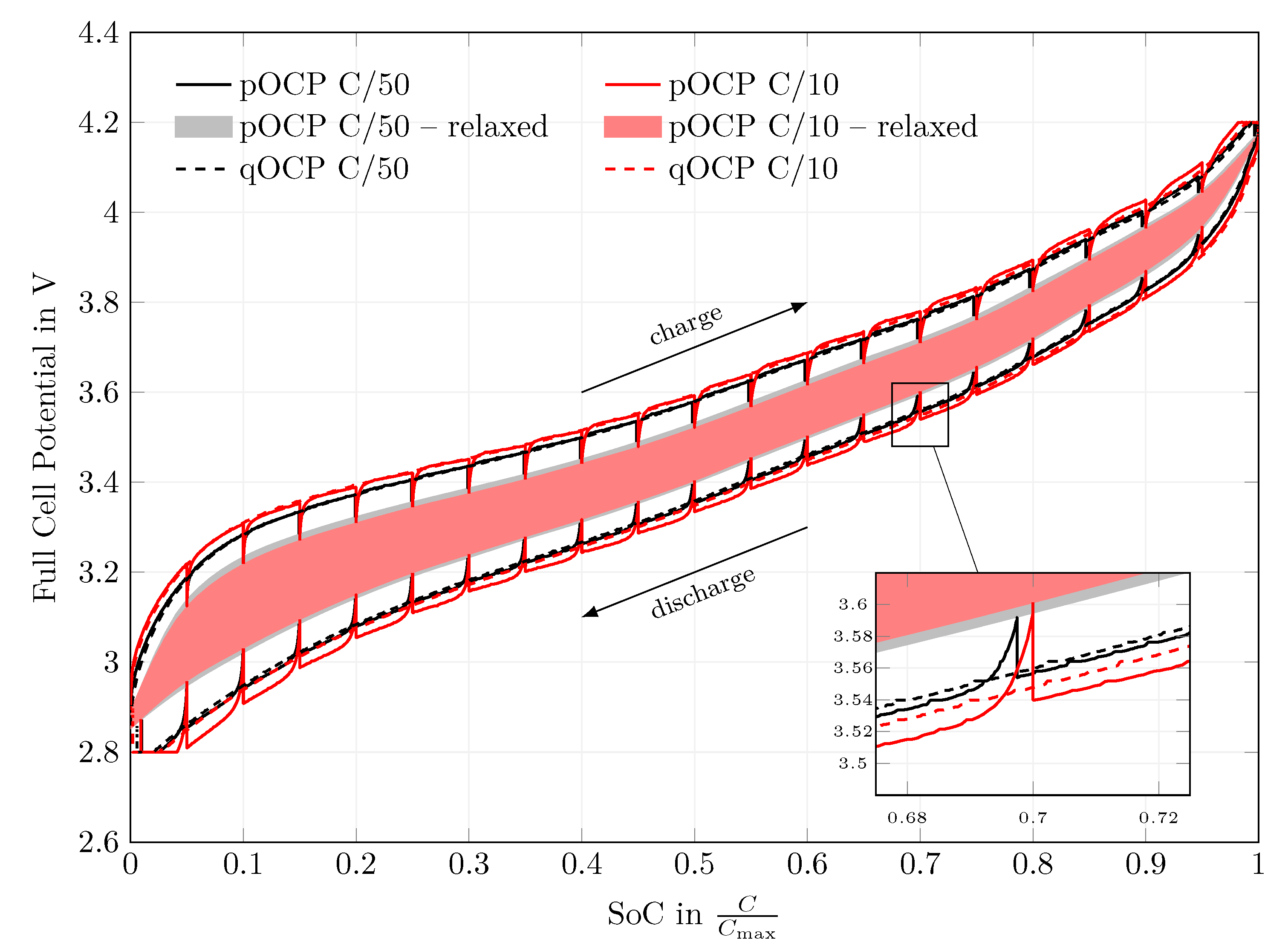
The cells then underwent one formation and nine stabilization cycles. Subsequently, a qOCP measurement at and was conducted for both coin and T-cells. The formation and stabilization cycles can be evaluated to retrieve an estimate of the capacity losses during formation, while the qOCP measurement yields the usable capacity after formation. This information will later be used to perform an x-axis transformation. The qOCP test is a constant current (CC) charge/discharge cycle at low C-rates of and which typically is used to measure the open-circuit voltage of a battery. In addition to the qOCP measurement using CC phases only, a pulsed open-circuit potential (pOCP) measurement was conducted. The pOCP test also makes use of a CC charge/discharge cycle at low C-rates of and . However, current-carrying phases are interrupted at every 5% state of charge (SoC) and the cell is allowed to relax for 2 h. The cell voltage after this relaxation period at different SoC is interpolated to measure the open-circuit voltage of a battery. The tests are compared to one another in order to investigate the qOCP as well as the polarization behavior at low currents. All tests were carried out at 25 and are assumed to be isothermal because of the large thermal capacity of the cell housing.
Subsequent to the qOCP measurement, a rate capability test was conducted using the T-cells in order to later validate the model at higher C-rates. The procedure includes CC charging and discharging at different C-rates of
,
,
,
,
,
,
,
, and
. Constant voltage (CV) phases were used for preconditioning so that every (dis)charge step starts with defined and reproducible conditions. More detailed descriptions of the exact procedures can be found in
Section S3 along with
Table S3 in the Supplementary Materials.
5. Conclusions and Outlook
Until now, the literature has not provided an electrochemical model for a silicon-dominant electrode. Hence, the goal of this study was to parametrize and validate a Newman-type model for a custom-built SiNCA lithium-ion cell. Many parameters in the model presented here, such as the porosity, could be obtained from electrode manufacturing, while other parameters, such as the exchange current density, were taken from the literature and underwent a fitting procedure.
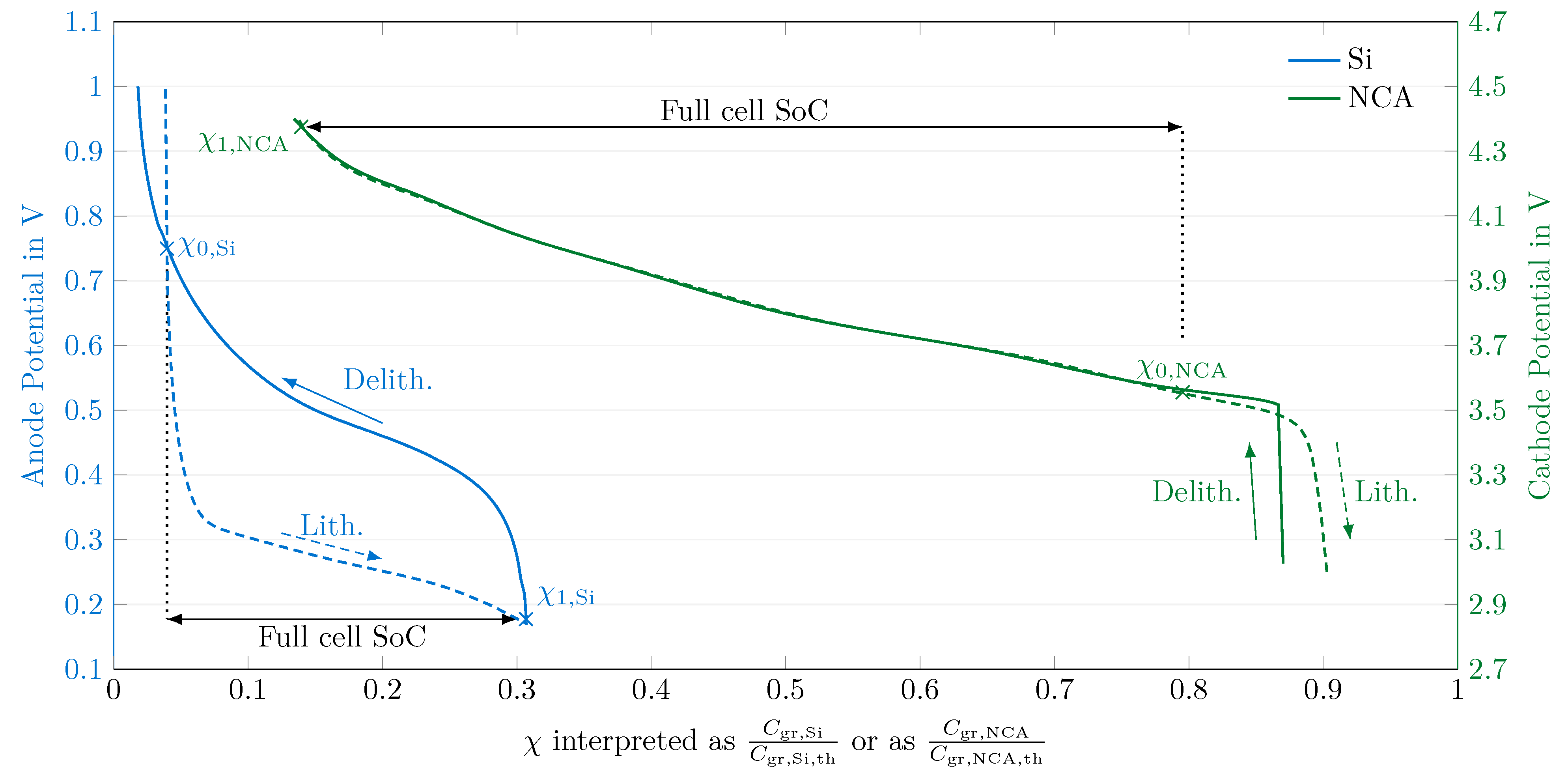
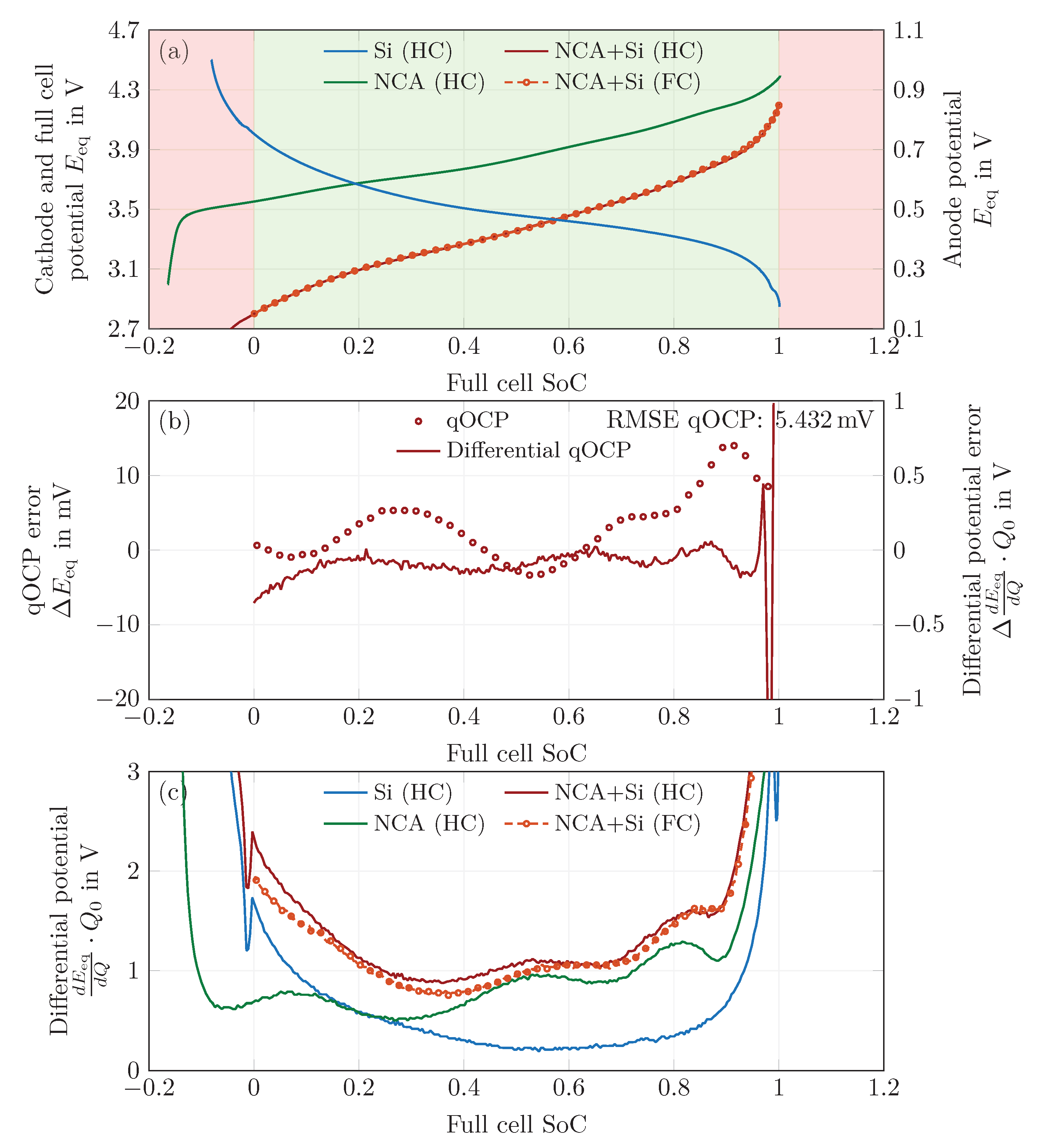
In a first step, the equilibrium potentials of the half cells and the full cell were measured. At C-rates as low as , the Si half-cell potential still relaxes significantly, but the origin of this effect has not been identified yet. The behavior at is similar to that at , indicating that more processes than just the charge transfer described by a single Butler–Volmer equation might be at play. Thus, the qOCP measurement at constant current have been used in the following, ignoring the model’s equilibrium potential relaxation effect. Voltage hysteresis is accounted for by switching between lithiation and delithiation half-cell potentials.
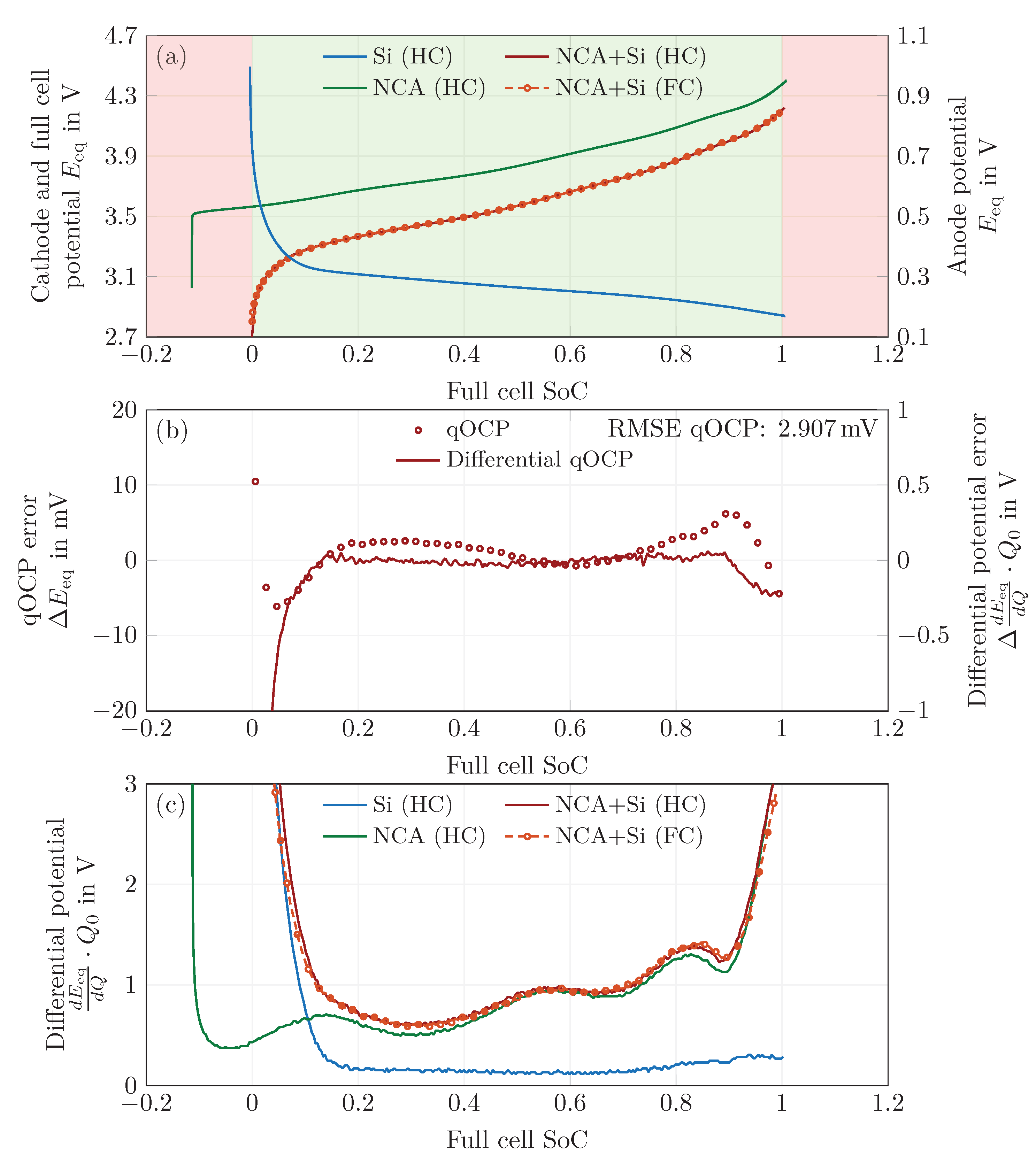
In a second step, a balancing process of the measured half cell qOCP curves was performed. A proper superposition of the half-cell potentials then yields the reconstructed full-cell qOCP curve with a RMSE of for the charge step and for the discharge step. The measurements needed for this balancing process also allow for an estimation of the anode and cathode utilization, and as such allow for an estimation of the DoL.
In a third step, the maximum solid-phase concentrations were fitted in such way that the model’s relative concentration is always bounded by the minimum and maximum DoL obtained in the previous step. Additionally, the concentrations were scaled so that the model’s capacity meets that of the measurements.
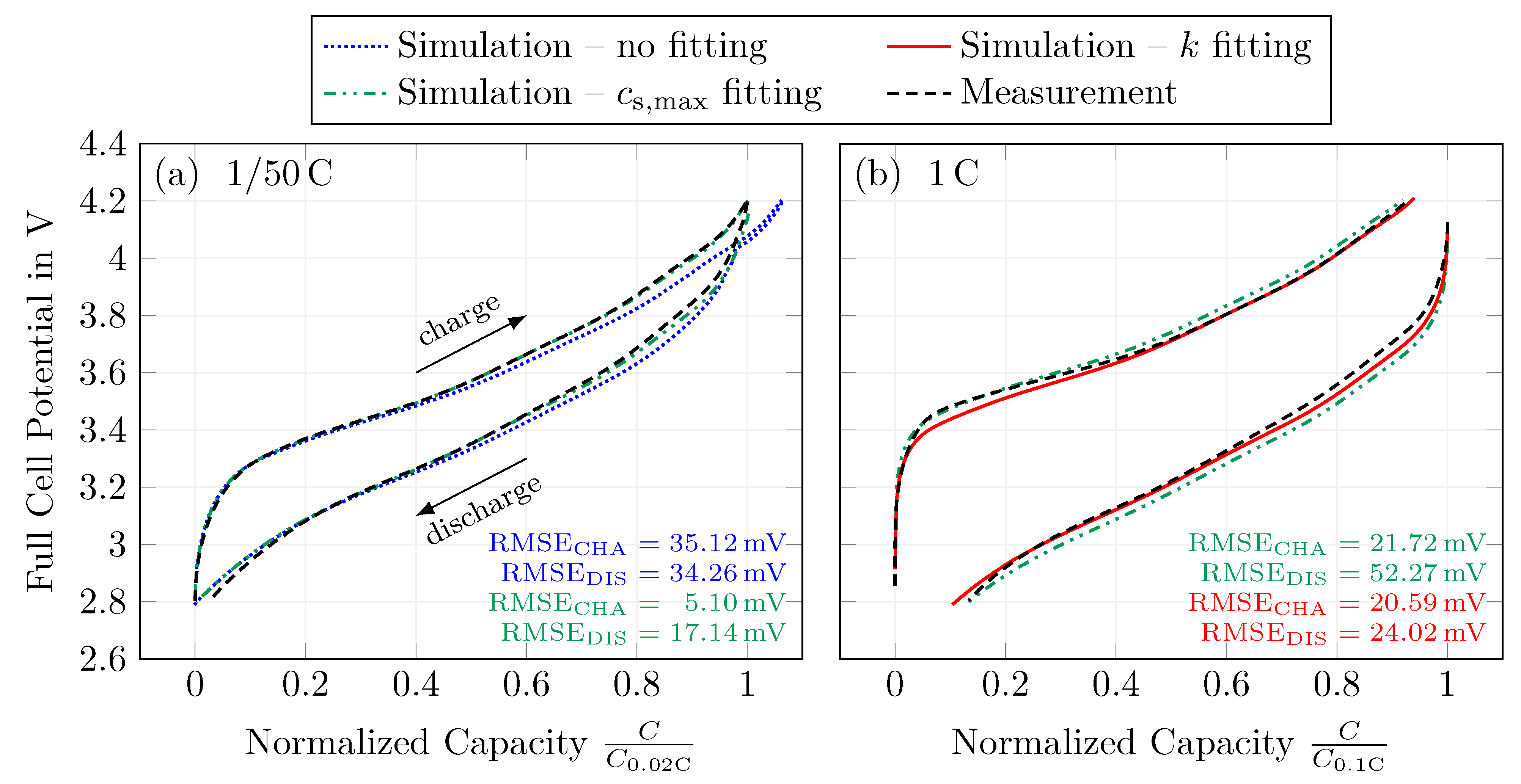
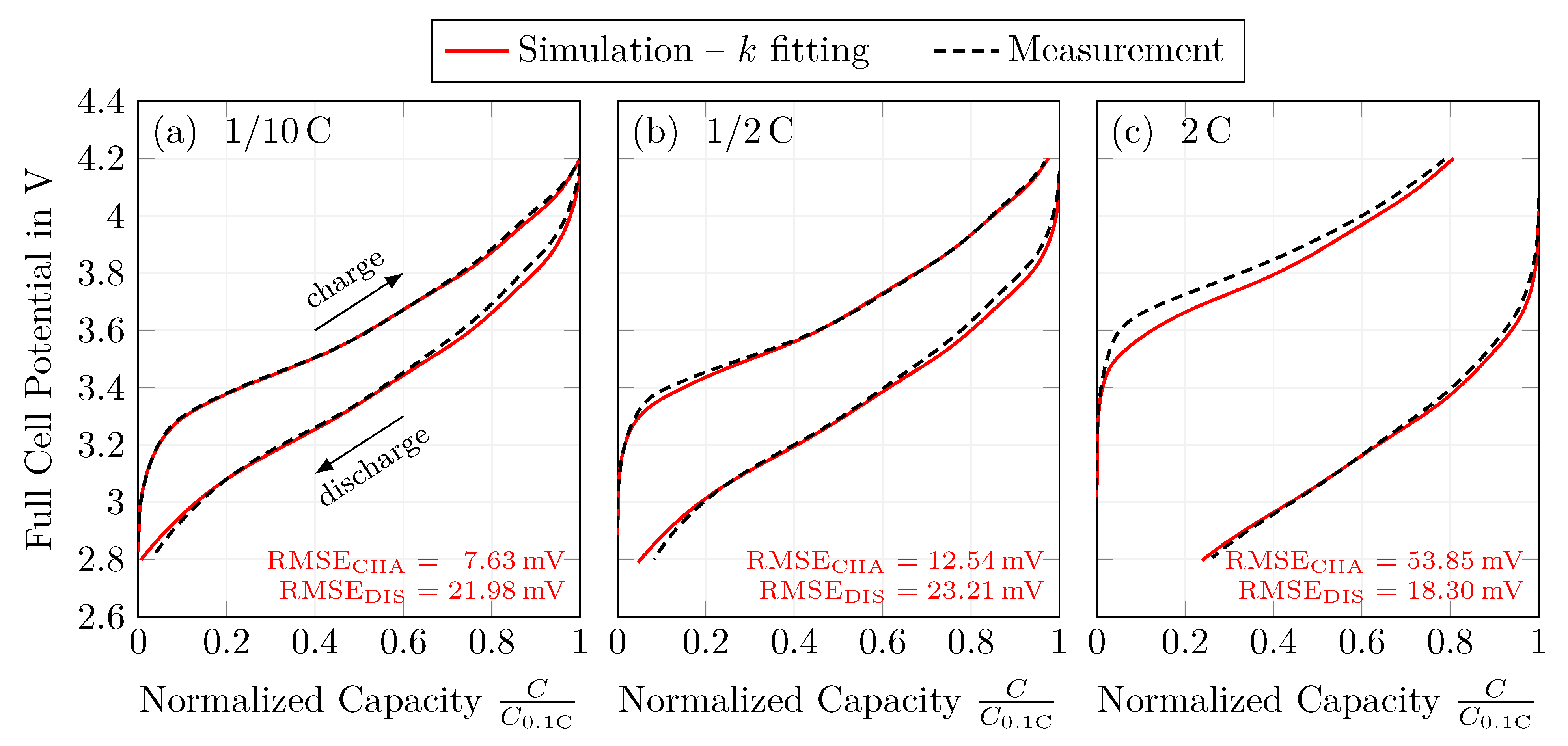
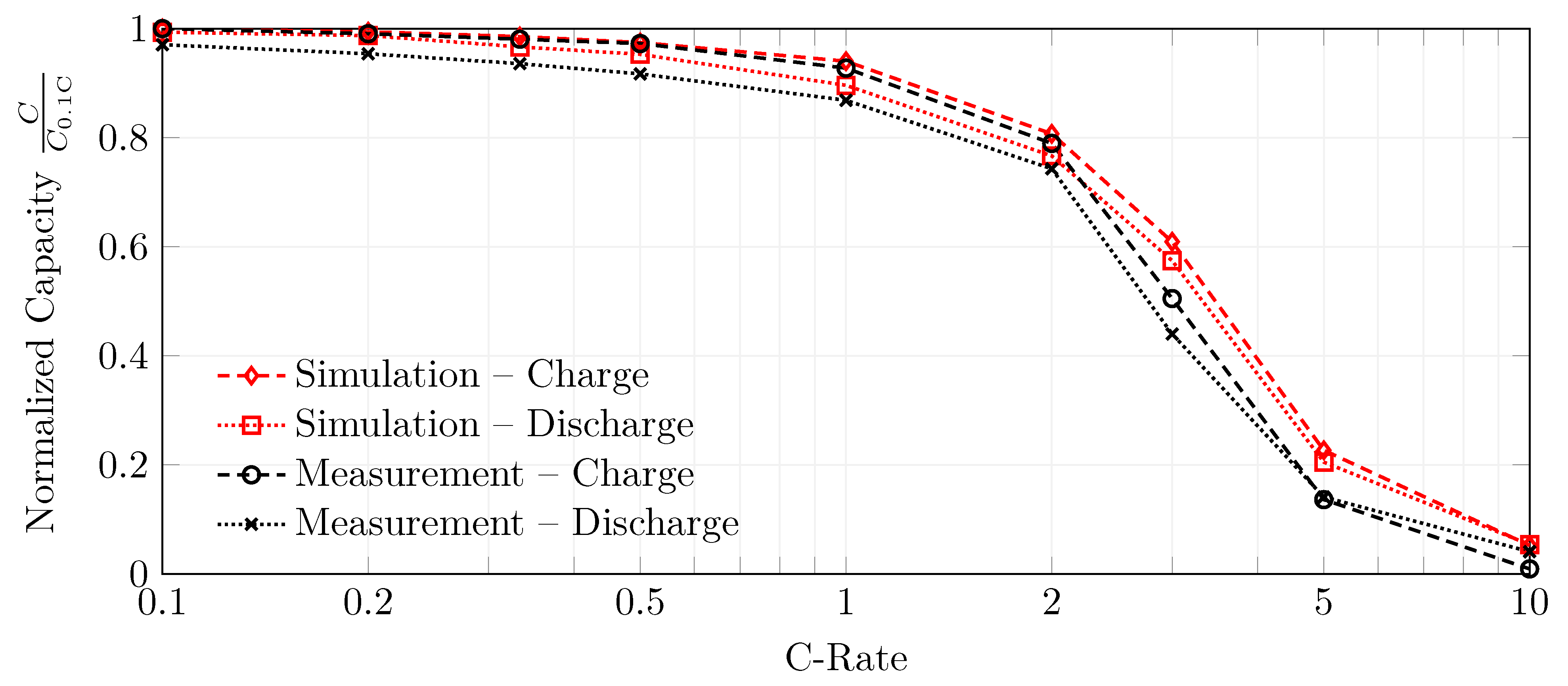
In a fourth and final step, the model’s reaction rate constants were fitted for a constant current charge and discharge step. The fitting was performed in such a way that the full-cell potential during both charging and discharging is described by a single parameter set. Different C-rates between and 10 were used to validate the model parametrization. The model can describe the full-cell potential and also meets the constant current (dis)charge capacity. This is true up to a C-rate of 2 . At higher C-rates, the model underestimates the polarization when comparing the simulation results with measurements.
Overall, the model results are satisfying, with an RMSE between measurements and the simulations of and during 1 charging and discharging, respectively. At the same time, the absolute error in the normalized capacities is 1.27% and 2.75%, respectively. Thus, the presented parametrization is considered valid to describe a silicon-dominant anode when a Newman-type model is used, under the premise that:
Si is partially lithiated to a similar degree as in this study and as such does not undergo further amorphization or crystallization.
Model equations have not been adjusted, for example with respect to complex electrode kinetics, solid state diffusion, or volumetric changes.
Voltage hysteresis is accounted for via two equilibrium curves rather than a dedicated hysteresis model.
In future work, several aspects regarding silicon-dominant anodes can be further investigated. Optimized parameters such as DoL-dependencies for the solid-phase diffusivity or the electrical conductivity could further improve the model quality. Additionally, a dedicated voltage hysteresis model could prove valuable to better describe the voltage behavior during open-circuit periods. An overview, including suggestions for experimental studies, is given for each of the parameters that have been taken from literature. An understanding of the origin of the strong relaxation behavior, including an appropriate kinetics model, would benefit subsequent studies. Closely related to this is an analysis of polarization at high currents. Since only laboratory cells were used, isothermal conditions were assumed for all experiments and simulations. A scale-up to a large-format pouch cell, for example, would require a thermal model to capture temperature-related effects such as joule heating and entropic heating. Similarly, a suitable mechanical model, because of the large volumetric expansion, is needed to capture porosity changes and related effects on the mass transport. Eventually, a fully coupled physicochemical-mechanical-thermal model would be required to capture all mentioned effects properly.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





