

Comparative Transcriptome Analysis Reveals Differential Gene Expression Pattern Associated with Heat Tolerance in Pepper (Capsicum annuum L.)
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Treatments
2.2. RNA Extraction, Library Construction, and RNA Sequencing
2.3. Quality Control and Read Mapping
2.4. Functional Enrichment Analysis
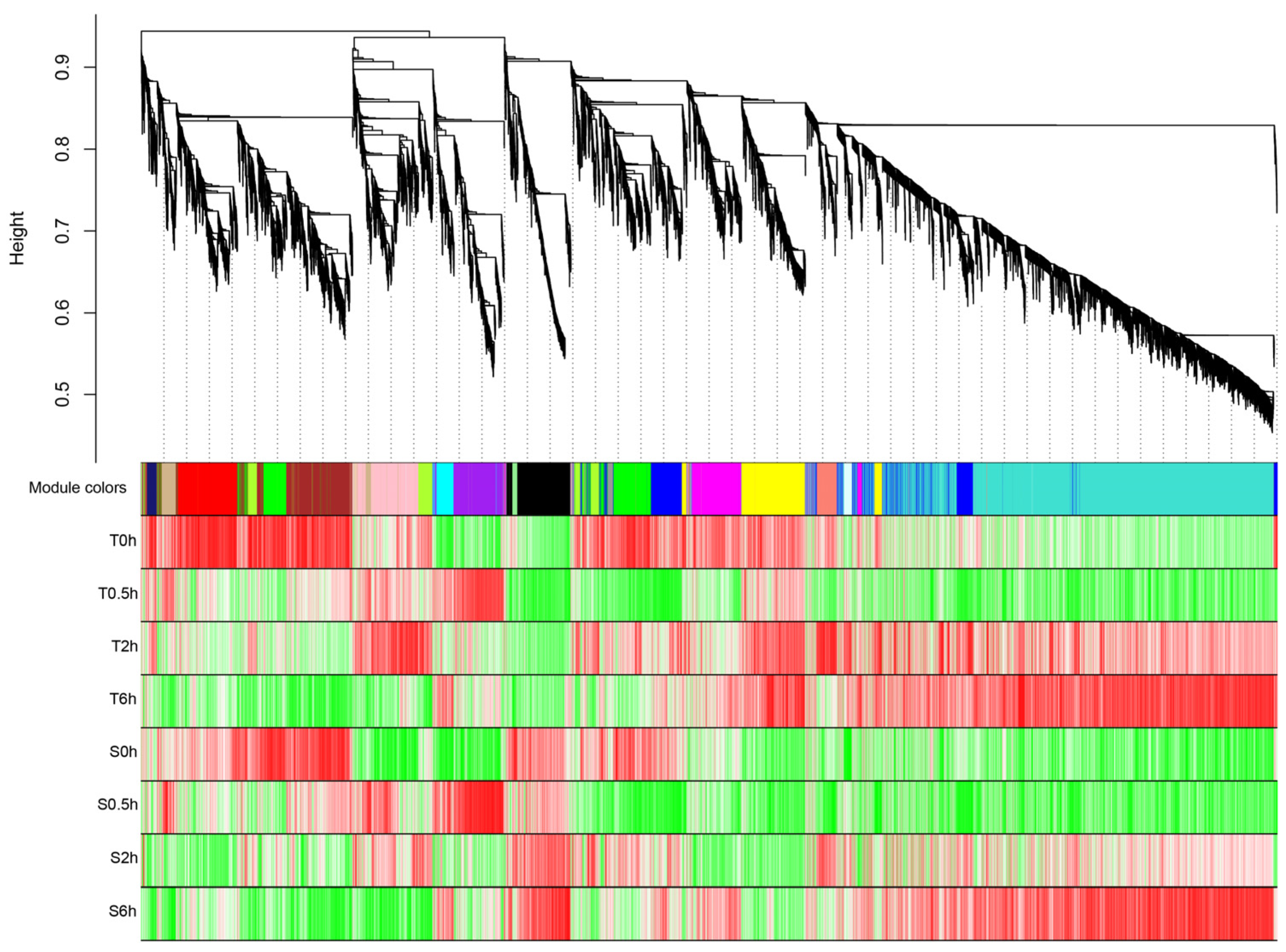
2.5. Construction of WGCNA
2.6. Quantitative Real-Time PCR
3. Results
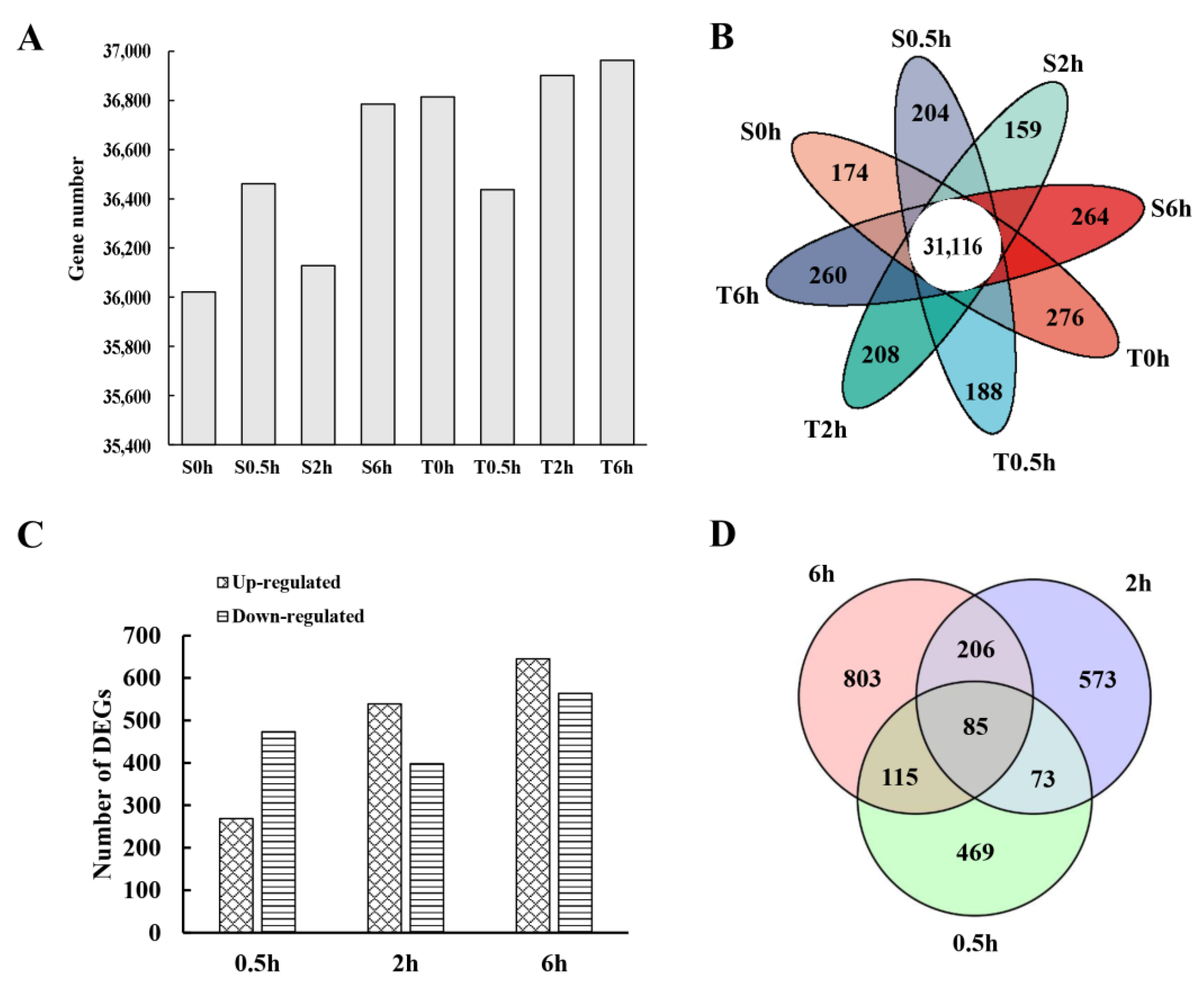
3.1. RNA Sequencing and DEGs Identification
3.2. Key Genes Screening for Heat Tolerance Based on WGCNA Analysis
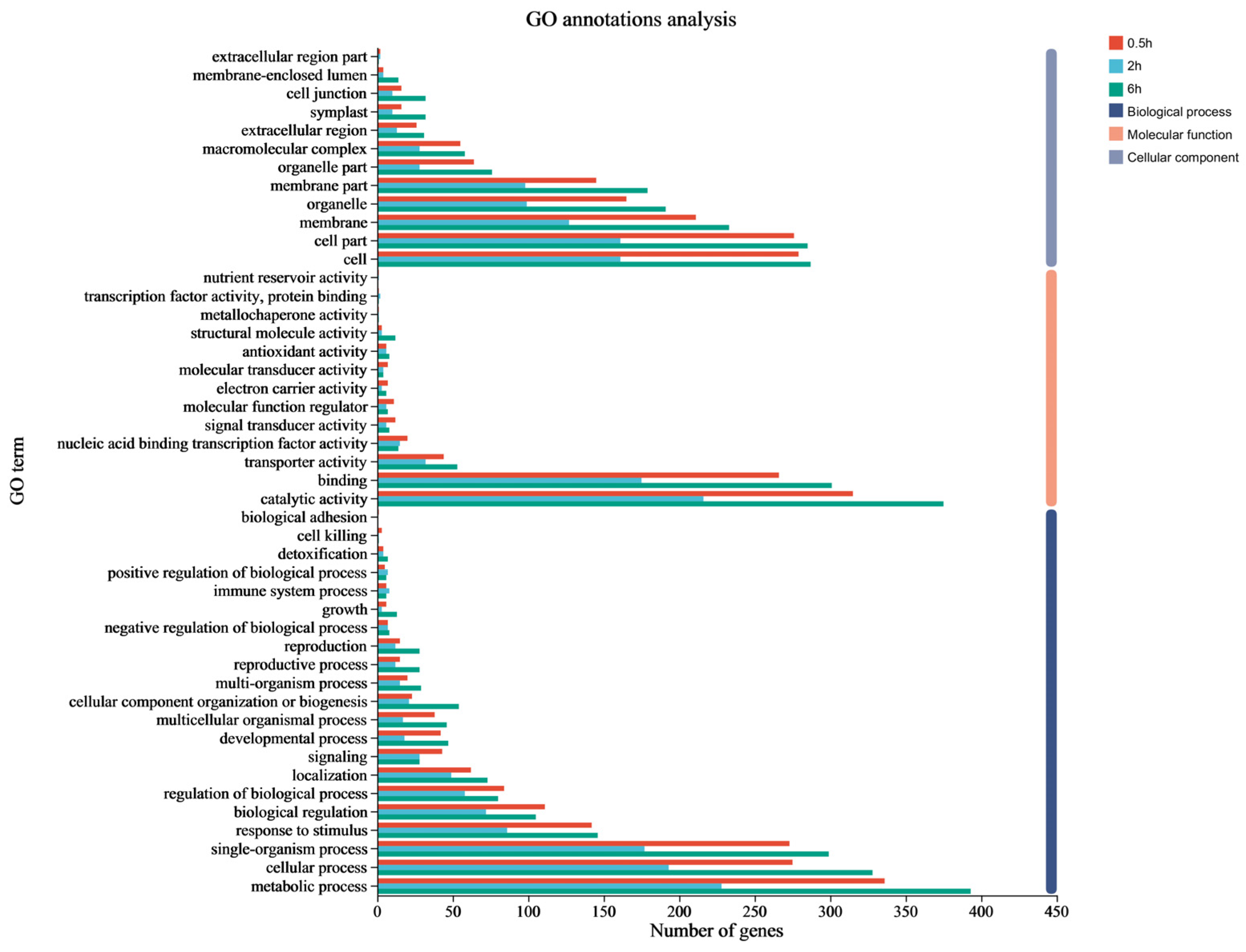
3.3. Functional Annotation by GO
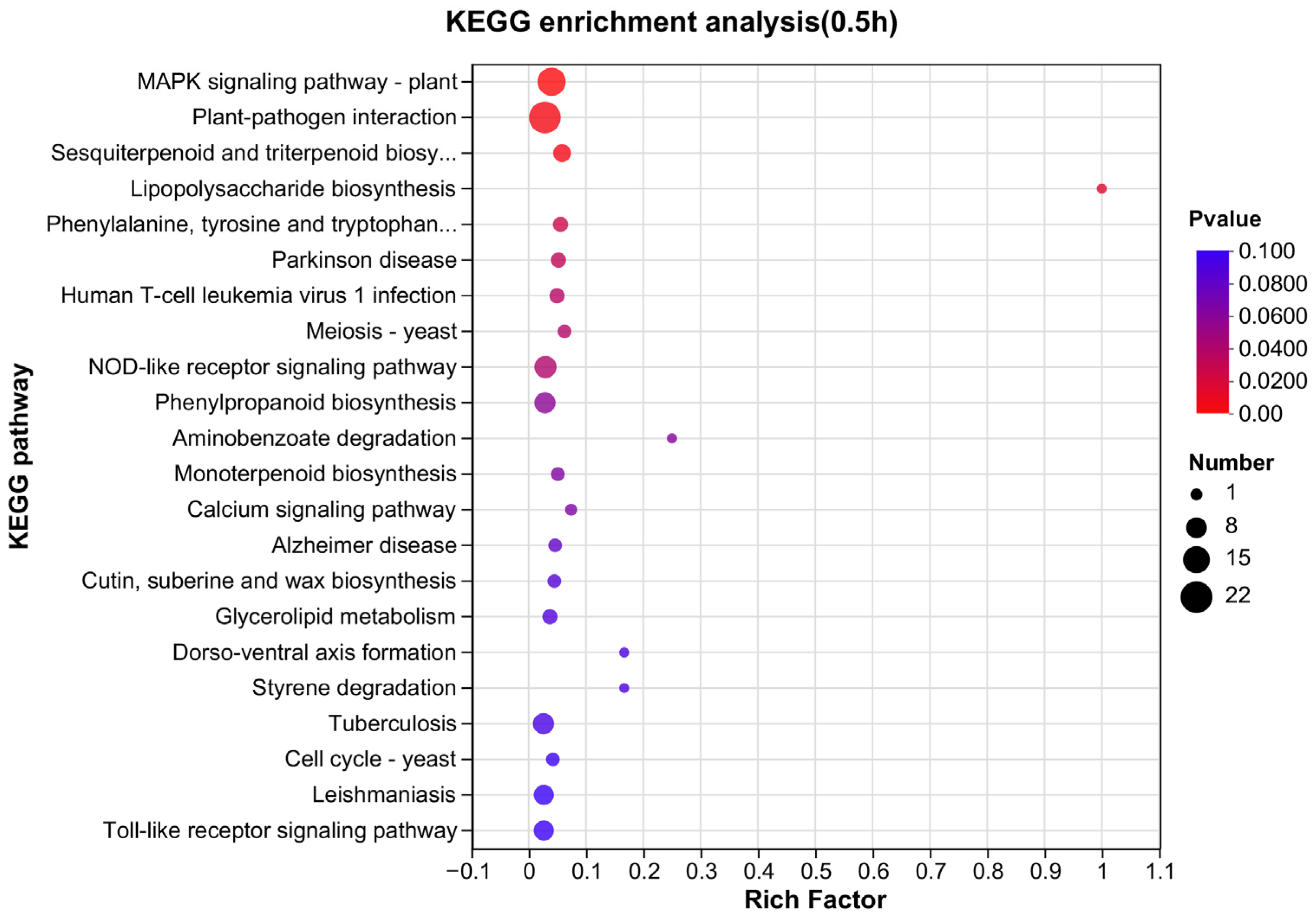
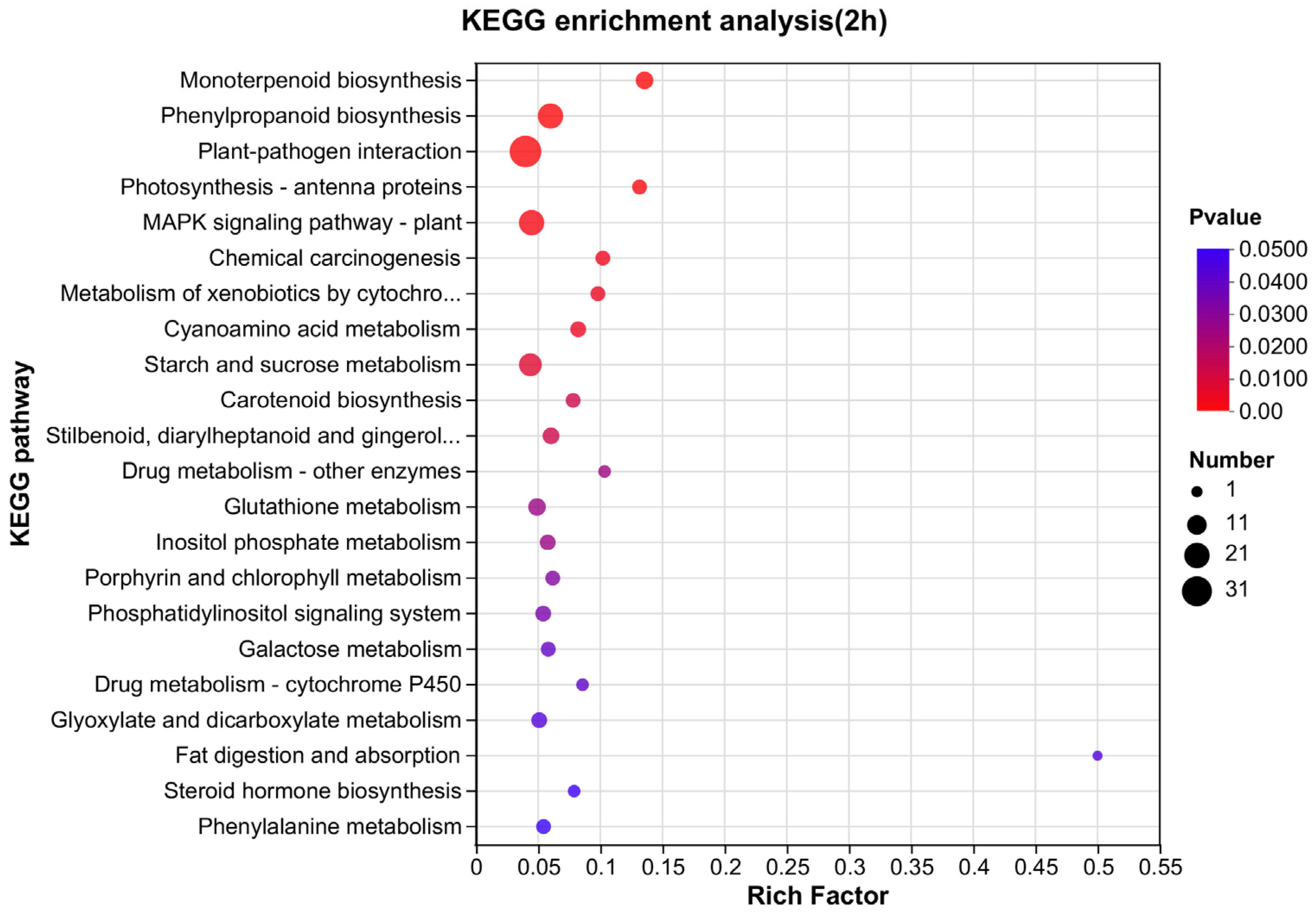
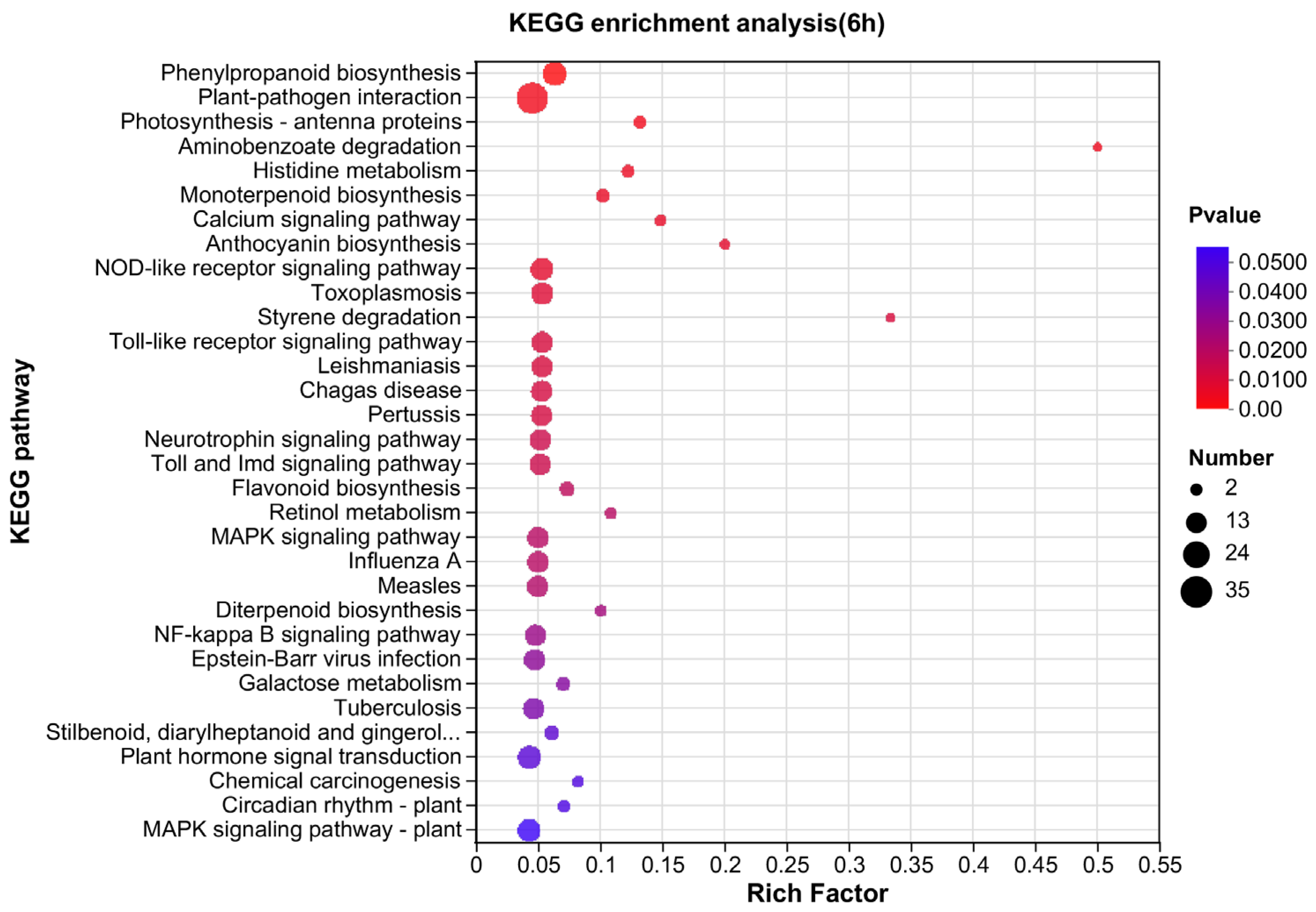
3.4. Pathway Mapping by KEGG
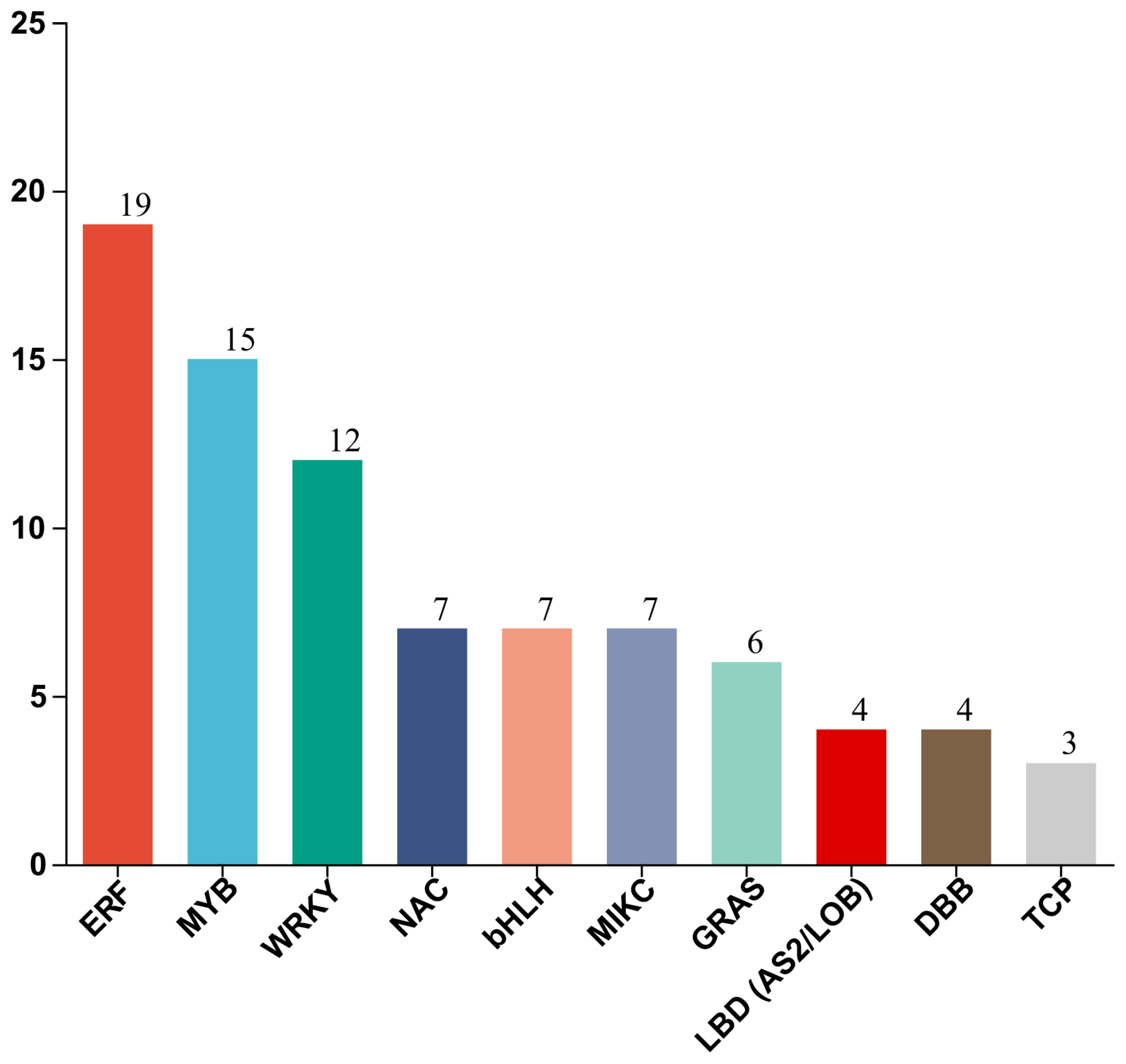
3.5. Identification of Differentially Expressed TFs
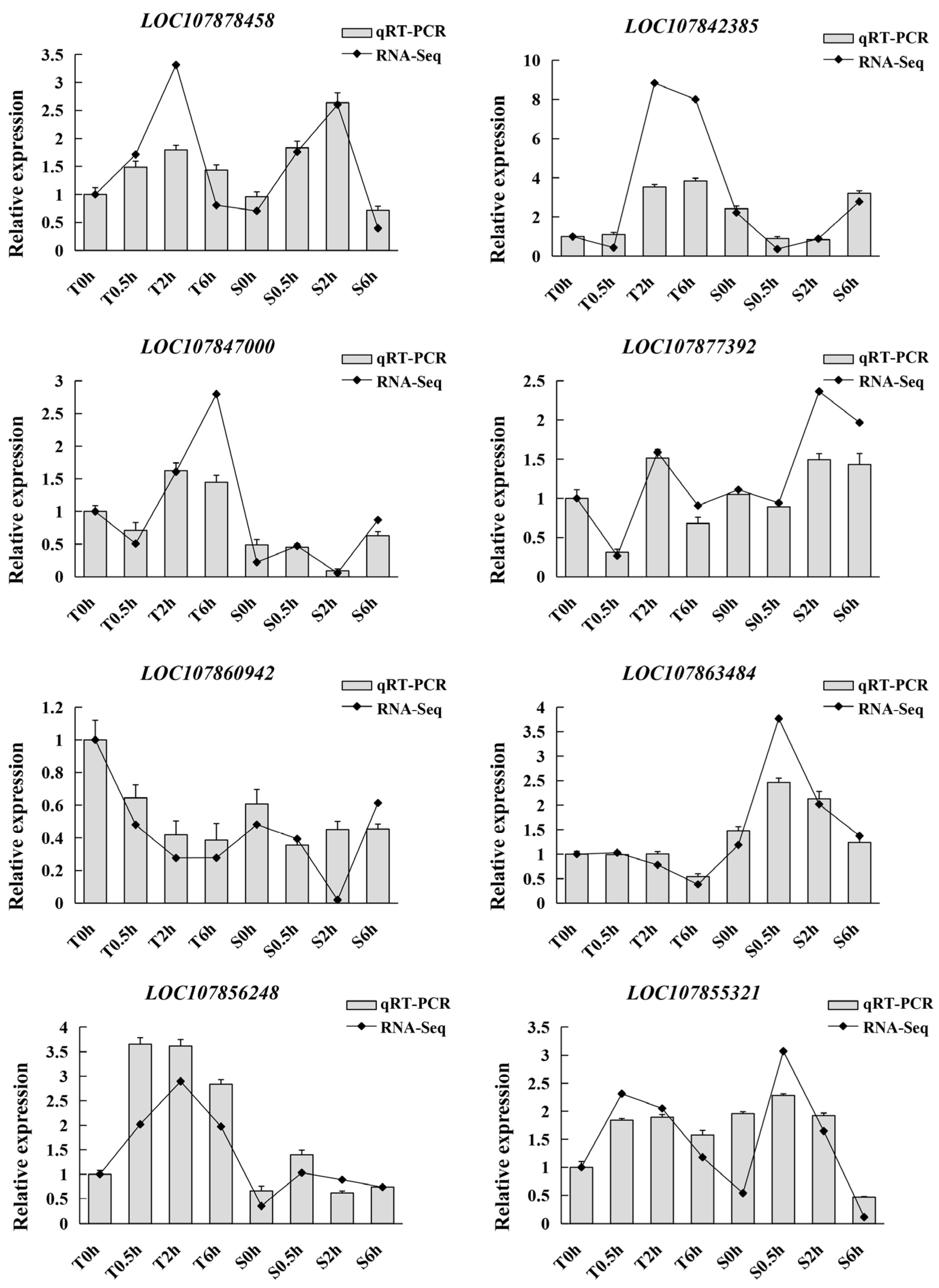
3.6. Validation of RNA-Seq Data by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Olatunji, T.L.; Afolayan, A.J. The suitability of chilli pepper (Capsicum annuum L.) for alleviating human micronutrient dietary deficiencies: A review. Food Sci. Nutr. 2018, 6, 2239–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.G.; Kim, S.K.; Lee, H.J.; Lee, H.S.; Lee, J.H. Impact of moderate and extreme climate change scenarios on growth, morphological features, photosynthesis, and fruit production of hot pepper. Ecol. Evol. 2018, 8, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Li, X.; Zhang, X.; Yin, Q.; Xie, L.; Zou, X.; Liu, F.; Dai, X. Transcriptome data reveal gene clusters and key genes in pepper response to heat shock. Front. Plant Sci. 2022, 13, 946475. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.M.; Zhou, X.; Chen, C.M.; Chen, C.J.; Chen, K.H.; Chen, M.X.; Zhu, Z.S. Coexpression network analysis reveals an MYB transcriptional activator involved in capsaicinoid biosynthesis in hot peppers. Hortic. Res. 2020, 7, 162. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yin, Y.; Yu, C.; Li, N.; Shen, S.; Liu, Y.; Gao, S.; Jiao, C.; Yao, M. Transcriptomics analysis of heat stress-induced genes in pepper (Capsicum annuum L.) seedlings. Horticulturae 2021, 7, 339. [Google Scholar] [CrossRef]
- Guo, M.; Zhai, Y.F.; Lu, J.P.; Chai, L.; Chai, W.G.; Gong, Z.H.; Lu, M.H. Characterization of CaHsp70-1, a pepper heat-shock protein gene in response to heat stress and some regulation exogenous substances in Capsicum annuum L. Int. J. Mol. Sci. 2014, 15, 19741–19759. [Google Scholar] [CrossRef] [Green Version]
- Saidi, Y.; Finka, A.; Muriset, M.; Bromberg, Z.; Weiss, Y.G.; Maathuis, F.J.; Goloubinoff, P. The heat shock response in moss plants is regulated by specific calcium-permeable channels in the plasma membrane. Plant Cell 2009, 21, 2829–2843. [Google Scholar] [CrossRef] [Green Version]
- Haider, S.; Iqbal, J.; Naseer, S.; Yaseen, T.; Shaukat, M.; Bibi, H.; Ahmad, Y.; Daud, H.; Abbasi, N.L.; Mahmood, T. Molecular mechanisms of plant tolerance to heat stress: Current landscape and future perspectives. Plant Cell Rep. 2021, 40, 2247–2271. [Google Scholar] [CrossRef]
- Nadeem, M.; Li, J.; Wang, M.; Shah, L.; Lu, S.; Wang, X.; Ma, C. Unraveling field crops sensitivity to heat stress: Mechanisms, approaches, and prospects. Agronomy 2018, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zang, X.; Kabir, M.R.; Liu, K.; Liu, Z.; Ni, Z.; Yao, Y.; Hu, Z.; Sun, Q.; Peng, H. A wheat lipid transfer protein 3 could enhance Arabidopsis’s basal thermotolerance and oxidative stress resistance. Gene 2014, 550, 18–26. [Google Scholar] [CrossRef]
- Kong, F.; Wang, J.; Cheng, L.; Liu, S.; Wu, J.; Peng, Z.; Lu, G. Genome-wide analysis of the mitogen-activated protein kinase gene family in Solanum lycopersicum. Gene 2012, 499, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, V.; Örvar, B.L.; Beyerly, J.; Hirt, H.; Dhindsa, R.S. Opposite changes in membrane fluidity mimic cold and heat stress activation of distinct plant MAP kinase pathways. Plant J. 2002, 31, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, S. Mitogen-activated protein kinase cascades in signalling plant growth and development. Trends Plant Sci. 2015, 20, 56–64. [Google Scholar] [CrossRef]
- Ding, H.; He, J.; Wu, Y.; Wu, X.; Ge, C.; Wang, Y.; Zhong, S.; Peiter, E.; Liang, J.; Xu, W. The tomato mitogen-activated protein kinase SlMPK1 is a negative regulator of the high-temperature stress response. Plant Physiol. 2018, 177, 633–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Qin, K.; Song, X.; Zhang, Q.; Zhou, Y.; Xia, X.; Yu, J. BZR1 transcription factor regulates heat stress tolerance through FERONIA Receptor-Like Kinase-Mediated reactive oxygen species signalling in tomato. Plant Cell Physiol. 2018, 59, 2239–2254. [Google Scholar] [PubMed]
- Liu, H.C.; Charng, Y. Common and distinct functions of Arabidopsis class A1 and A2 heat shock factors in diverse abiotic stress responses and development. J. Plant Physiol. 2013, 163, 276–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, M.; Liu, H.J.; Ma, X.; Zhen, H.G.; Lu, M.H. The plant heat stress transcription factors (HSFs): Structure, regulation, and function in response to abiotic stresses. Front. Plant Sci. 2016, 7, 114. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, Y.; Maruyama, K.; Qin, F.; Osakabe, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Dual function of an Arabidopsis transcription factor DREB2A in water-stress-responsive and heat-stress-responsive gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18822–18827. [Google Scholar] [CrossRef]
- Konzen, E.R.; Recchia, G.H.; Cassieri, F.; Caldas, D.G.G.; Berny Mier, Y.; Teran, J.C.; Gepts, P.; Tsai, S.M. DREB genes from common bean (Phaseolus vulgaris) show broad to specific abiotic stress responses and distinct levels of nucleotide diversity. Int. J. Genom. 2019, 2019, 9520642. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Mizoi, J.; Tanaka, H.; Maruyama, K.; Qin, F.; Osakabe, Y.; Morimoto, K.; Ohori, T.; Kusakabe, K.; Nagata, M.; et al. Arabidopsis DPB3-1, a DREB2A interactor, specifically enhances heat stress-induced gene expression by forming a heat stress-specific transcriptional complex with NF-Y subunits. Plant Cell 2014, 26, 4954–4973. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved transcriptome reconstruction from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A Python library for Gene Ontology analyses. Sci. Rep. 2018, 18, 10872. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Chen, J.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, S.; Sidhu, G.P.S. Climate change regulated abiotic stress mechanisms in plants: A comprehensive review. Plant Cell Rep. 2022, 41, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, J.; He, N.; Guo, F. Metabolic reprogramming in chloroplasts under heat stress in plants. Int. J. Mol. Sci. 2018, 19, 849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Yan, B.; Lou, X.; Ma, H.; Ruan, S. Comparative transcriptome analysis reveals the transcriptional alterations in heat-resistant and heat-sensitive sweet maize (Zea mays L.) varieties under heat stress. BMC Plant Biol. 2017, 17, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Luo, S.; Zhao, Y.; Miao, Y.; Wang, Q.; Ye, L.; Gao, L.; Ahammed, G.J.; Cheng, Y. Multiomics analyses reveal high temperature-induced molecular regulation of ascorbic acid and capsaicin biosynthesis in pepper fruits. Environ. Exp. Bot. 2022, 201, 104941. [Google Scholar] [CrossRef]
- Kuang, J.; Wu, C.; Guo, Y.; Walther, D.; Shan, W.; Chen, J.; Chen, L.; Lu, W. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis. Plant Biotechnol. J. 2021, 19, 477–489. [Google Scholar] [CrossRef]
- Greenham, K.; Guadagno, C.R.; Gehan, M.A.; Mockler, T.C.; Weinig, C.; Ewers, B.E.; McClung, C.R. Temporal network analysis identifies early physiological and transcriptomic indicators of mild drought in Brassica rapa. eLife 2017, 6, e29655. [Google Scholar] [CrossRef]
- Wang, J.; Lv, J.; Liu, Z.; Liu, Y.; Song, J.; Ma, Y.; Ou, L.; Zhang, X.; Liang, C.; Wang, F.; et al. Integration of transcriptomics and metabolomics for pepper (Capsicum annuum L.) in response to heat stress. Int. J. Mol. Sci. 2019, 20, 5042. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Chattopadhyay, S. Glutathione modulates the expression of heat shock proteins via the transcription factors BZIP10 and MYB21 in Arabidopsis. J. Exp. Bot. 2018, 69, 3729–3743. [Google Scholar] [CrossRef] [Green Version]
- Latijnhouwers, M.; Xu, X.M.; Møller, S.G. Arabidopsis stromal 70-kDa heat shock proteins are essential for chloroplast development. Planta 2010, 232, 567–578. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, Z.; Wang, L.; Jin, B. Plant responses to heat stress: Physiology, transcription, noncoding RNAs, and epigenetics. Int. J. Mol. Sci. 2020, 22, 117. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.; Gamage, D.; Hirotsu, N.; Martin, A.; Seneweera, S. Effects of elevated carbon dioxide on photosynthesis and carbon partitioning: A perspective on root sugar sensing and hormonal crosstalk. Front. Physiol. 2017, 8, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arce-Rodriguez, M.L.; Ochoa-Alejo, N. Biochemistry and molecular biology of capsaicinoid biosynthesis: Recent advances and perspectives. Plant Cell Rep. 2019, 38, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Luan, Y.; Meng, J.; Sun, J.; Tao, J.; Zhao, D. WRKY transcription factor response to high-temperature stress. Plants 2021, 10, 2211. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.K.; Tripp, J.; Winkelhaus, S.; Tschiersch, B.; Theres, K.; Nover, L.; Scharf, K.D. In the complex family of heat stress transcription factors, HsfA1 has a unique role as master regulator of thermotolerance in tomato. Genes Dev. 2002, 16, 1555–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Fu, Q.; Huang, W.; Yu, D. Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep. 2009, 28, 683–693. [Google Scholar] [CrossRef]
- Arce-Rodriguez, M.L.; Ochoa-Alejoa, N. An R2R3-MYB transcription factor regulates capsaicinoid biosynthesis. Plant Physiol. 2017, 174, 1359–1370. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Zhu, Z.; Liu, K.; Zhao, Y.; Nie, Z.; Zhang, L.; Muhammad Fahim, A.; Yang, X. Comparative Transcriptome Analysis Reveals Differential Gene Expression Pattern Associated with Heat Tolerance in Pepper (Capsicum annuum L.). Horticulturae 2023, 9, 801. https://doi.org/10.3390/horticulturae9070801
Song Y, Zhu Z, Liu K, Zhao Y, Nie Z, Zhang L, Muhammad Fahim A, Yang X. Comparative Transcriptome Analysis Reveals Differential Gene Expression Pattern Associated with Heat Tolerance in Pepper (Capsicum annuum L.). Horticulturae. 2023; 9(7):801. https://doi.org/10.3390/horticulturae9070801
Chicago/Turabian StyleSong, Yunpeng, Zongwen Zhu, Kaige Liu, Yuelu Zhao, Zhixing Nie, Lili Zhang, Abbas Muhammad Fahim, and Xuedong Yang. 2023. "Comparative Transcriptome Analysis Reveals Differential Gene Expression Pattern Associated with Heat Tolerance in Pepper (Capsicum annuum L.)" Horticulturae 9, no. 7: 801. https://doi.org/10.3390/horticulturae9070801