
Genetic Diversity and Population Structure Analysis of Hollyhock (Alcea rosea Cavan) Using High-Throughput Sequencing
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Analysis of Phenotypic Traits
2.3. DNA Extraction, Sequencing, and SNP Genotyping
2.4. Genetic Diversity and Population Structure Analysis
3. Results
3.1. Phenotypic Variations of Six Qualitative Traits
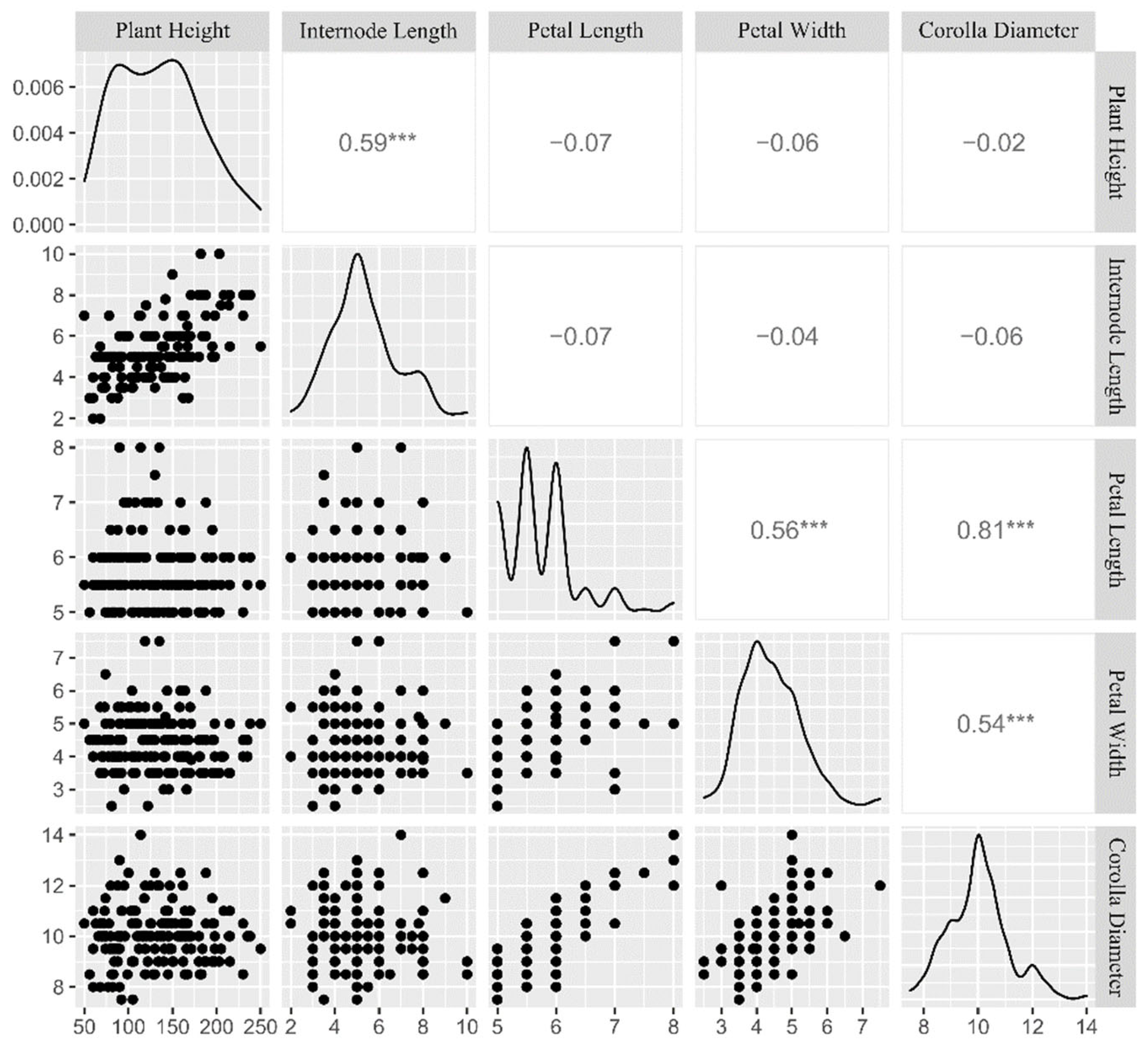
3.2. Phenotypic Variation and Correlation Analyses among Five Quantitative Traits
3.3. Single-Nucleotide Polymorphism Detection and Nucleotide Diversity
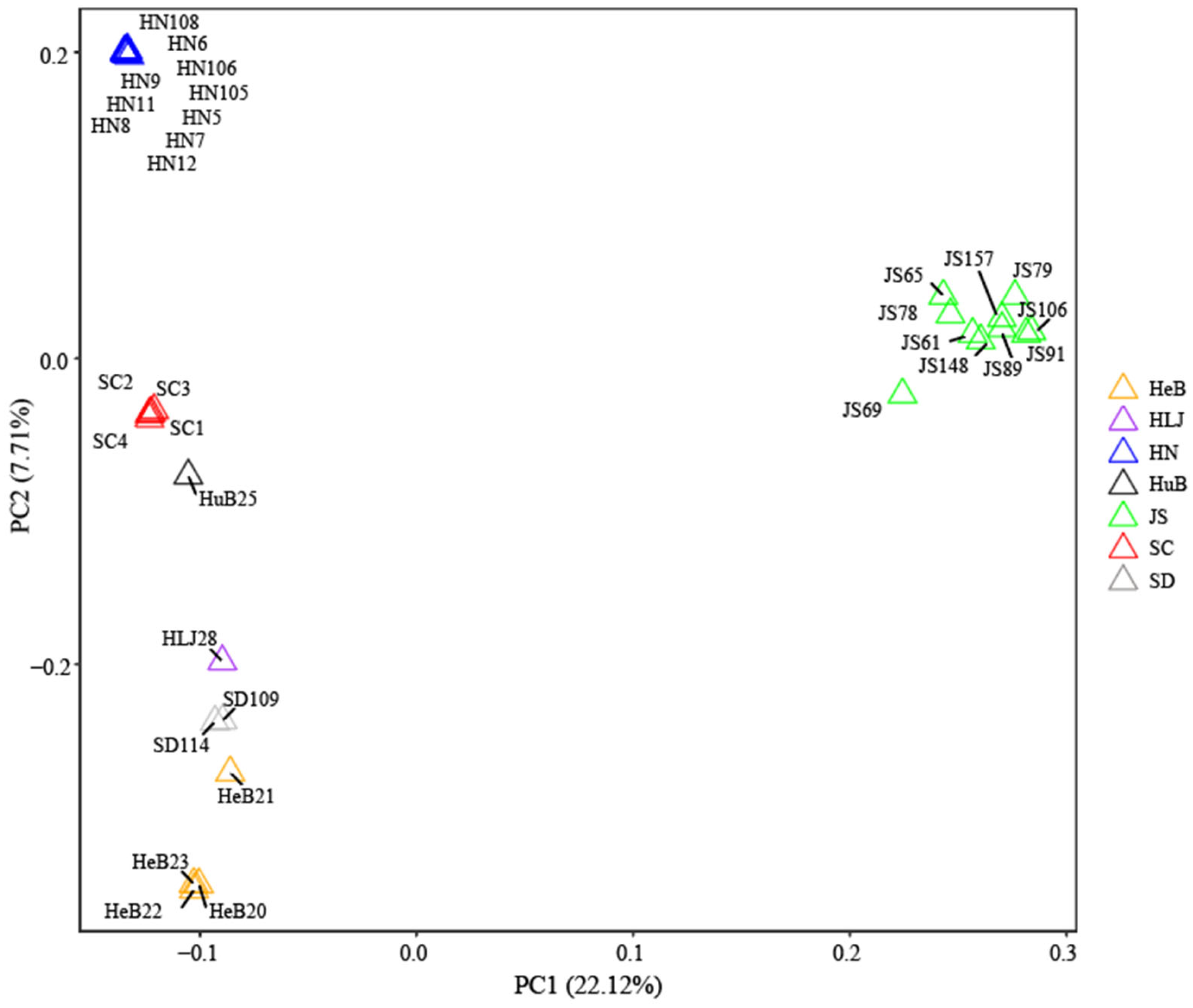
3.4. Principal Component Analysis of Hollyhock Accessions
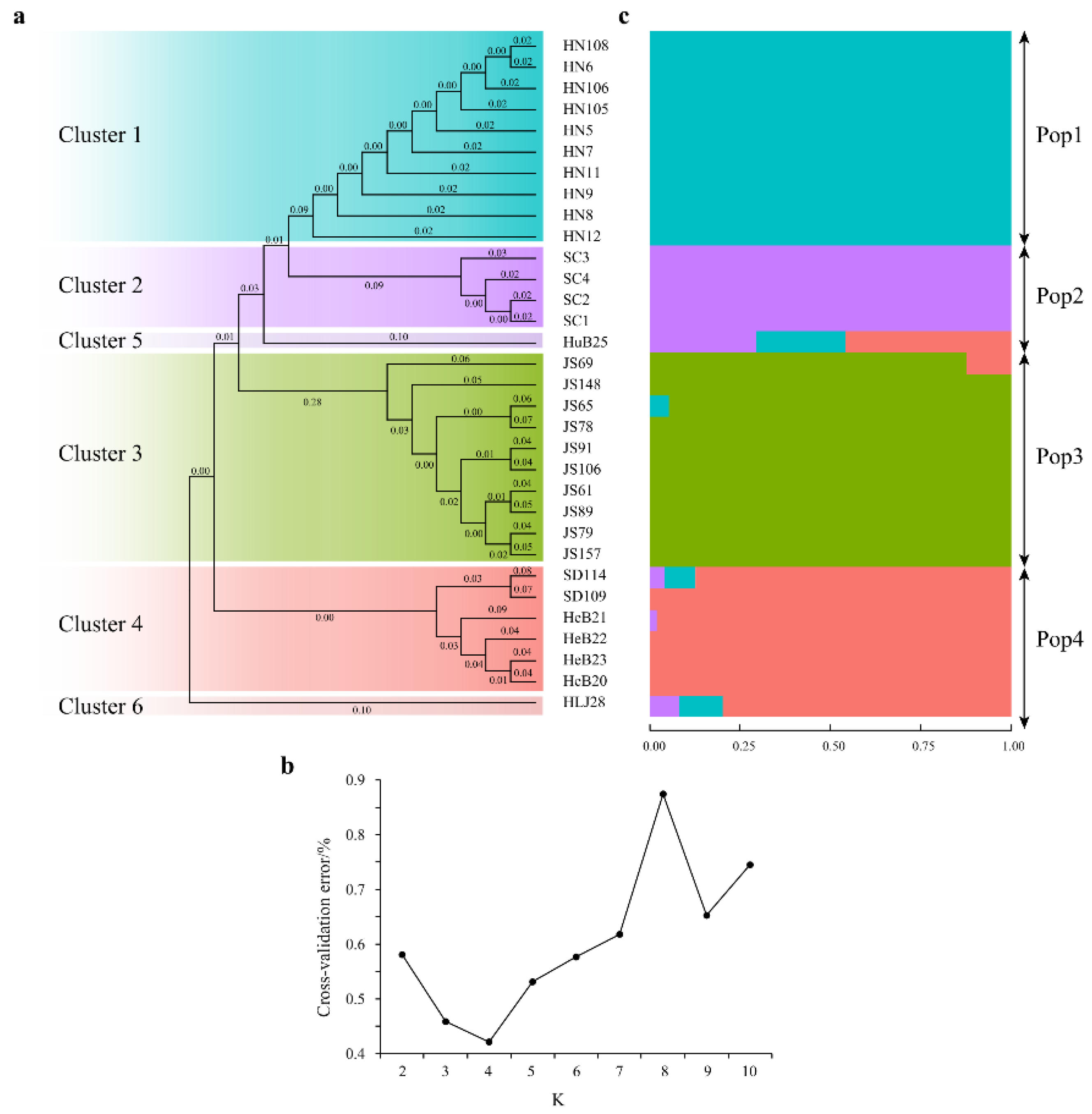
3.5. Phylogenetic Tree and Population Structure Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.; Lan, Y.; Mi, R.; Xu, M.; Liu, Y.; Wu, J.; Zhang, Y.; Long, L.; Yang, G. Effects of intercropping ryegrass with hollyhock and inoculation with Bacillus thuringiensis on enhancing phytoremediation of Cd-contaminated soils. Chemosphere 2023, 311, 136974. [Google Scholar] [CrossRef]
- Shehzad, M.R.; Hanif, M.A.; Rehman, R.; Bhatti, I.A. Hollyhock. In Medicinal Plants of South Asia: Novel Sources for Drug Discovery, 1st ed.; Muhammad, A.H., Haq, N., Muhammad, M.K., Hugh, J.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 381–391. [Google Scholar]
- Li, S. Compendium of Materia Medica; People’s Medical Publishing House: Beijing, China, 1982. [Google Scholar]
- Munir, M.; Hussain, A.; Ul-Haq, I.; Qureshi, R.; Munazir, M.; Arshad, M.N.; Leghari, M.K. Callogenesis potential of cotyledonary explants of Althaea rose L. from Pakistan. Pak. J. Bot. 2012, 44, 271–275. [Google Scholar]
- Wang, S. A Study of the Paintings of Xixia Grottoes in Dunhuang on the background of art history. J. Dunhuang Stud. 2019, 104, 153. [Google Scholar]
- Yi, C. A study on Xia Hua Tu by Li Song in Song Dynasty. Master’s Thesis, Zhejiang Nnomal University, Zhejiang, China, 2022. [Google Scholar]
- Gokhale, S.B.; Tatiya, A.U.; Bakliwal, S.; Fursule, R.A. Natural dye yielding plants. Indian J. Nat. Prod. Resour. 2004, 3, 228–234. [Google Scholar]
- Ahmadi, M.; Rad, A.K.; Rajaei, Z.; Hadjzadeh, M.A.; Mohammadian, N.; Tabasi, N.S. Alcea rosea root extract as a preventive and curative agent in ethylene glycol-induced urolithiasis in rats. Indian J. Pharmacol. 2012, 44, 304–307. [Google Scholar] [CrossRef]
- Lim, T.K. Edible Medicinal and Non-Medicinal Plants, 1st ed.; Springer: Canberra, Australia, 2012; p. 835. [Google Scholar]
- Zhang, Y.; Jin, L.; Chen, Q.; Wu, Z.; Dong, Y.; Han, L.; Wang, T. Hypoglycemic activity evaluation and chemical study on hollyhock flowers. Fitoterapia 2015, 102, 7–14. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, E.K.; Nawarathna, W.; Dong, X.; Shin, W.B.; Park, J.S.; Moon, S.H.; Park, P.J. Immune-Stimulatory Effects of Althaea rosea Flower Extracts through the MAPK Signaling Pathway in RAW264.7 Cells. Molecules 2017, 22, 679. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Zu, L.; Zhang, M.; Yang, T.; Zhou, M.; Shi, C.; Shi, F.; Zhang, W. Tolerance and distribution of cadmium in an ornamental species Althaea rosea Cavan. Int. J. Phytoremediat. 2020, 22, 713–724. [Google Scholar] [CrossRef]
- Liu, J.N.; Zhou, Q.X.; Sun, T. Growing responses and hyperaccumulating characteristics of three ornamental plants to Cd-Pb combined pollution. Acta Sci. Circumstantiae 2006, 26, 2039–2044. [Google Scholar]
- Liu, J.; Zhou, Q.; Wang, S.; Sun, T. Cadmium tolerance and accumulation of Althaea rosea Cav. and its potential as a hyperaccumulator under chemical enhancement. Environ. Monit. Assess. 2009, 149, 419–427. [Google Scholar] [CrossRef]
- Janfaza, S.; Janfaza, E. The study of medicinal plants usage trough urban green space. Ann. Biol. Res. 2012, 3, 1934–1937. [Google Scholar]
- Leus, L.; Van Laere, K.; De Riek, J.; Van Huylenbroeck, J. Rose. In Ornamental Crops, 1st ed.; Van Huylenbroeck, J., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 719–767. [Google Scholar]
- Marasek-Ciolakowska, A.; Nishikawa, T.; Shea, D.J.; Okazaki, K. Breeding of lilies and tulips—Interspecific hybridization and genetic background. Breed. Sci. 2018, 68, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Marasek-Ciolakowska, A.; Sochacki, D.; Marciniak, P. Breeding Aspects of Selected Ornamental Bulbous Crops. Agronomy 2021, 11, 1709. [Google Scholar] [CrossRef]
- Xing, G.; Qu, L.; Zhang, W.; Zhang, Y.; Yuan, X.; Lei, J. Study on interspecific hybridization between tulip cultivars and wild species native to China. Euphytica 2020, 216, 66. [Google Scholar] [CrossRef]
- Yu, S.; Sun, W.; Wang, J.; Li, Z. Rice germplasm and its pivotal role in functional genomics research. Chin. Bull. Life Sci. 2016, 28, 1122–1128. [Google Scholar] [CrossRef]
- He, T.; Li, C. Harness the power of genomic selection and the potential of germplasm in crop breeding for global food security in the era with rapid climate change. Crop. J. 2020, 8, 688–700. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.; Li, S.; Chen, Q.; Teixeira da Silva, J.A.; Wang, A.; Yu, X.; Wang, L. Germplasm resources and genetic breeding of Paeonia: A systematic review. Hortic. Res. 2020, 7, 107. [Google Scholar] [CrossRef]
- Sharma, S.; Schulthess, A.W.; Bassi, F.M.; Badaeva, E.D.; Neumann, K.; Graner, A.; Özkan, H.; Werner, P.; Knüpffer, H.; Kilian, B. Introducing Beneficial Alleles from Plant Genetic Resources into the Wheat Germplasm. Biology 2021, 10, 982. [Google Scholar] [CrossRef]
- Roy, R.; Sultana, S.; Begum, N.; Fornara, D.; Barmon, M.; Zhang, R.; Sarker, T.; Rabbany, M.G. Exogenous melatonin reduces water deficit-induced oxidative stress and improves growth performance of Althaea rosea grown on coal mine spoils. Environ. Sci. Pollut. Res. 2022, 29, 61550–61560. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids. Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [Green Version]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.; Messina, C.D. Breeding crops for drought-affected environments and improved climate resilience. Plant Cell 2023, 35, 162–186. [Google Scholar] [CrossRef]
- Dwivedi, S.L.; Ceccarelli, S.; Blair, M.W.; Upadhyaya, H.D.; Are, A.K.; Ortiz, R. Landrace Germplasm for Improving Yield and Abiotic Stress Adaptation. Trends Plant Sci. 2016, 21, 31–42. [Google Scholar] [CrossRef]
- Eltaher, S.; Sallam, A.; Belamkar, V.; Emara, H.A.; Nower, A.A.; Salem, K.F.M.; Poland, J.; Baenziger, P.S. Genetic Diversity and Population Structure of F3:6 Nebraska Winter Wheat Genotypes Using Genotyping-By-Sequencing. Front. Genet. 2018, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Alemu, A.; Feyissa, T.; Letta, T.; Abeyo, B. Genetic diversity and population structure analysis based on the high density SNP markers in Ethiopian durum wheat (Triticum turgidum ssp. durum). BMC Genet. 2020, 21, 18. [Google Scholar] [CrossRef] [Green Version]
- Motahari, B.; Shabanian, N.; Rahmani, M.S.; Mohammad, H.F. Genetic diversity and genetic structure of Acer monspessulanum L. across Zagros forests of Iran using molecular markers. Gene 2021, 769, 145245. [Google Scholar] [CrossRef]
- Hao, C.Y.; Zhang, X.Y.; Wang, L.F.; Dong, Y.S.; Shang, X.W.; Jia, J.Z. Genetic Diversity and Core Collection Evaluations in Common Wheat Germplasm from the Northwestern Spring Wheat Region in China. Mol. Breed. 2006, 17, 69–77. [Google Scholar] [CrossRef]
- Wang, H.; Khera, P.; Huang, B.; Yuan, M.; Katam, R.; Zhuang, W.; Harris, S.K.; Moore, K.M.; Culbreath, A.K.; Zhang, X.; et al. Analysis of genetic diversity and population structure of peanut cultivars and breeding lines from China, India and the US using simple sequence repeat markers. J. Integr. Plant Biol. 2016, 58, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.J.; Oliver, M.; Gómez, P.H.; Vicente, M.C. Comparing AFLP, RAPD and RFLP markers for measuring genetic diversity in melon. Theor. Appl. Genet. 2000, 101, 860–864. [Google Scholar] [CrossRef]
- Pandey, A.; Khan, M.K.; Isik, R.; Turkmen, O.; Ramazan, A.; Musa, S.; Hakki, E.E. Genetic diversity and population structure of watermelon (Citrullus sp.) genotypes. 3 Biotech 2019, 9, 210. [Google Scholar] [CrossRef]
- Loera, S.M.; Studer, B.; Kölliker, R. DNA-Based Assessment of Genetic Diversity in Grassland Plant Species: Challenges, Approaches, and Applications. Agronomy 2019, 9, 881. [Google Scholar] [CrossRef] [Green Version]
- Brant, J.B.; Gustavo, C.A.; Peter, M.G. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 1991, 196, 80–83. [Google Scholar] [CrossRef]
- DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: www.genome.gov/sequencingcostsdata (accessed on 17 May 2023).
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.; Rafal, M.G.; Bruce, B.; Michael, B.; Robert, B.; James, H.; Melissa, K.; Nicholas, L.; Lynda, P.; Romay, M.C.; et al. Genomic estimation of complex traits reveals ancient maize adaptation to temperate North America. Science 2017, 357, 512–515. [Google Scholar] [CrossRef] [Green Version]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.B.; Yang, G.H.; Wong, F.L.; Li, M.W.; He, W.M.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef]
- Wang, B.; Lin, Z.; Li, X.; Zhao, Y.; Zhao, B.; Wu, G.; Ma, X.; Wang, H.; Xie, Y.; Li, Q.; et al. Genome-wide selection and genetic improvement during modern maize breeding. Nat. Genet. 2020, 52, 565–571. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Z.; Bao, Z.; Li, H.; Lyu, Y.; Zan, Y.; Wu, Y.; Cheng, L.; Fang, Y.; Wu, K.; et al. Graph pangenome captures missing heritability and empowers tomato breeding. Nature 2022, 606, 527–534. [Google Scholar] [CrossRef]
- Edea, Z.; Dadi, H.; Kim, S.W.; Dessie, T.; Lee, T.; Kim, H.; Kim, J.J.; Kim, K.S. Genetic diversity, population structure and relationships in indigenous cattle populations of Ethiopia and Korean Hanwoo breeds using SNP markers. Front. Genet. 2013, 4, 35. [Google Scholar] [CrossRef] [Green Version]
- Meisner, J.; Albrechtsen, A. Inferring Population Structure and Admixture Proportions in Low-Depth NGS Data. Genetics 2018, 210, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, S.A.; Prasanna, B.M. Analysis of Genetic Diversity in Crop Plants—Salient Statistical Tools and Considerations. Crop. Sci. 2003, 43, 1235–1248. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yuan, M.; Tao, A.; Xu, J.; Lin, L.; Fang, P.; Qi, J. Genetic Structure and Relationship Analysis of an Association Population in Jute (Corchorus spp.) Evaluated by SSR Markers. PLoS ONE 2015, 10, e0128195. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Zhang, C.; Mu, J.; Zhang, H.; Yao, W.; Ding, X.; Ding, J.; Chang, Y. All-in-one sequencing: An improved library preparation method for cost-effective and high-throughput next-generation sequencing. Plant Methods 2020, 16, 74. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, C.; Wang, L.; Luo, M.; Zhang, P.; Wang, Y.; Malik, W.A.; Wang, Y.; Chen, P.; Qiu, X.; et al. A prolific and robust whole-genome genotyping method using PCR amplification via primer-template mismatched annealing. J. Integr. Plant Biol. 2023, 65, 633–645. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Trait Description (Grade) and Coding |
|---|---|
| Petal Color | 1 = white, 2 = yellow, 3 = pink, 4 = red, 5 = crimson |
| Flower Type | 1 = single flower, 2 = double flower |
| Petal Margin | 1 = bilobed, 2 = multilobed, 3 = serrated |
| Flower Center Color | 1 = white, 2 = red, 3 = yellow |
| Petal Structure | 1 = fused, 2 = unfused |
| Leaf Shape | 1 = cordate, 2 = palmate trilobed, 3 = palmate bifurcate, 4 = palmate trifurcate |
| Trait | Distribution of Frequency (%) | H′ | ||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| Petal Color | 22.2 | 5.6 | 29.6 | 11.1 | 31.5 | 1.46 |
| Flower Type | 54.3 | 45.7 | - | - | - | 0.70 |
| Petal Margin | 29.0 | 43.8 | 27.2 | - | - | 1.08 |
| Flower Center Color | 4.3 | 27.8 | 67.9 | - | - | 0.75 |
| Petal Structure | 80.9 | 19.1 | - | - | - | 0.49 |
| Leaf Shape | 3.1 | 4.3 | 52.5 | 40.1 | - | 0.95 |
| Trait | Max. | Min. | Mean | S.D. | C.V. (%) | H′ |
|---|---|---|---|---|---|---|
| Plant Height (cm) | 250.1 | 30.0 | 130.3 | 49.7 | 38.2 | 2.18 |
| Internode Length (cm) | 10.0 | 2.1 | 5.3 | 1.5 | 27.9 | 1.88 |
| Petal Length (cm) | 8.0 | 5.0 | 5.7 | 0.6 | 10.9 | 1.47 |
| Petal Width (cm) | 7.6 | 2.5 | 4.4 | 0.8 | 18.9 | 1.85 |
| Corolla Diameter (cm) | 14.0 | 7.5 | 10.0 | 1.1 | 11.2 | 1.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, S.; Chen, P.; Liu, Y.; Ma, Z.; Malik, W.A.; Zhu, Z.; Peng, Z.; Lu, H.; Chen, Y.; et al. Genetic Diversity and Population Structure Analysis of Hollyhock (Alcea rosea Cavan) Using High-Throughput Sequencing. Horticulturae 2023, 9, 662. https://doi.org/10.3390/horticulturae9060662
Wang Y, Zhao S, Chen P, Liu Y, Ma Z, Malik WA, Zhu Z, Peng Z, Lu H, Chen Y, et al. Genetic Diversity and Population Structure Analysis of Hollyhock (Alcea rosea Cavan) Using High-Throughput Sequencing. Horticulturae. 2023; 9(6):662. https://doi.org/10.3390/horticulturae9060662
Chicago/Turabian StyleWang, Yue, Sheng Zhao, Peng Chen, Yongkang Liu, Zhigang Ma, Waqar Afzal Malik, Zhenghang Zhu, Zhenyu Peng, Haorong Lu, Yanli Chen, and et al. 2023. "Genetic Diversity and Population Structure Analysis of Hollyhock (Alcea rosea Cavan) Using High-Throughput Sequencing" Horticulturae 9, no. 6: 662. https://doi.org/10.3390/horticulturae9060662