

Combined Study of Transcriptome and Metabolome Reveals Involvement of Metabolites and Candidate Genes in Flavonoid Biosynthesis in Prunus avium L.

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Extraction, Library Preparation
2.3. Bioinformatics Analysis of RNA-seq
2.4. Sample Preparation and Metabolite Extraction
2.5. Analyzing Wide-Target Metabolomics Data Qualitatively and Quantitatively
2.6. Data Processing and Analysis
3. Results
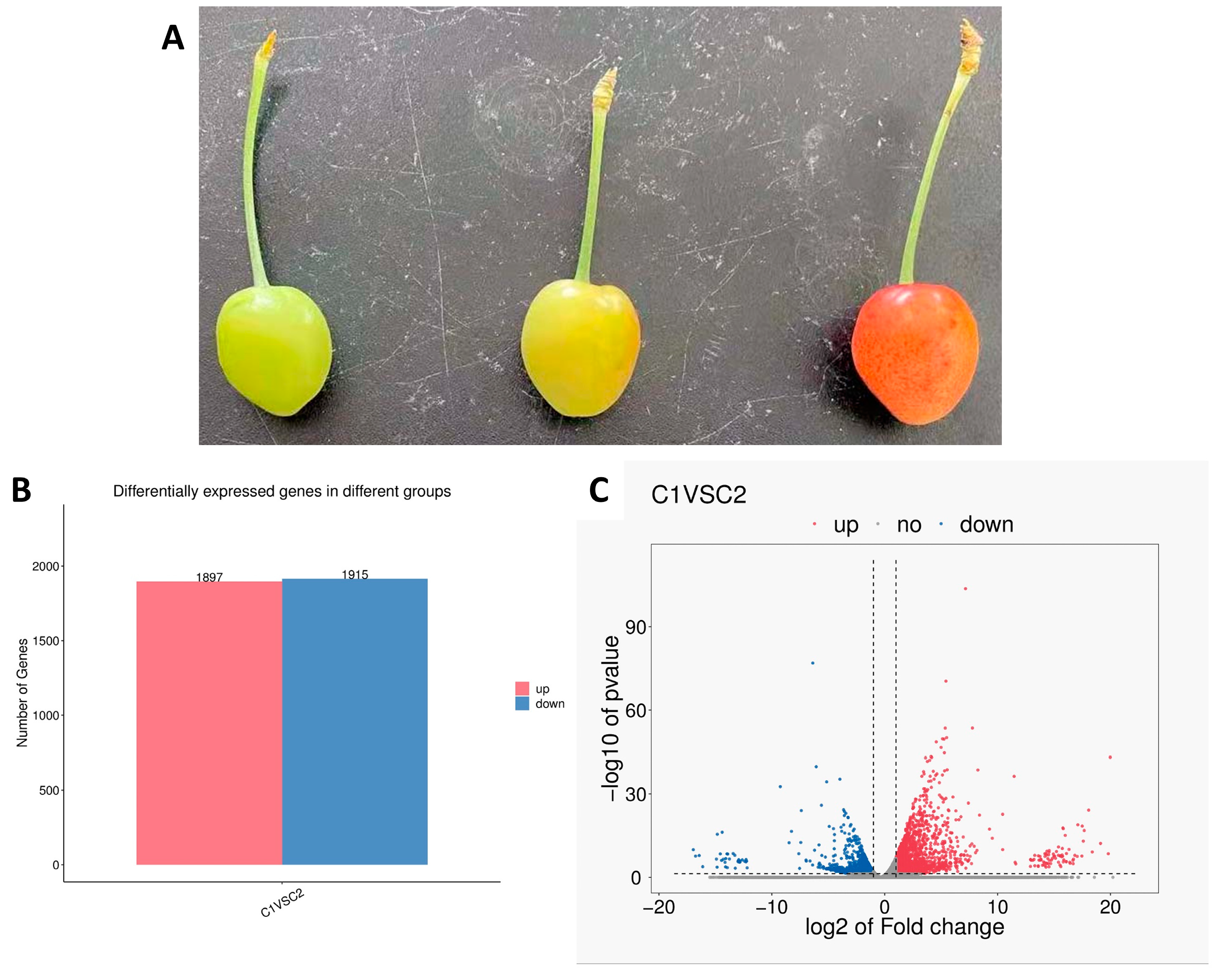
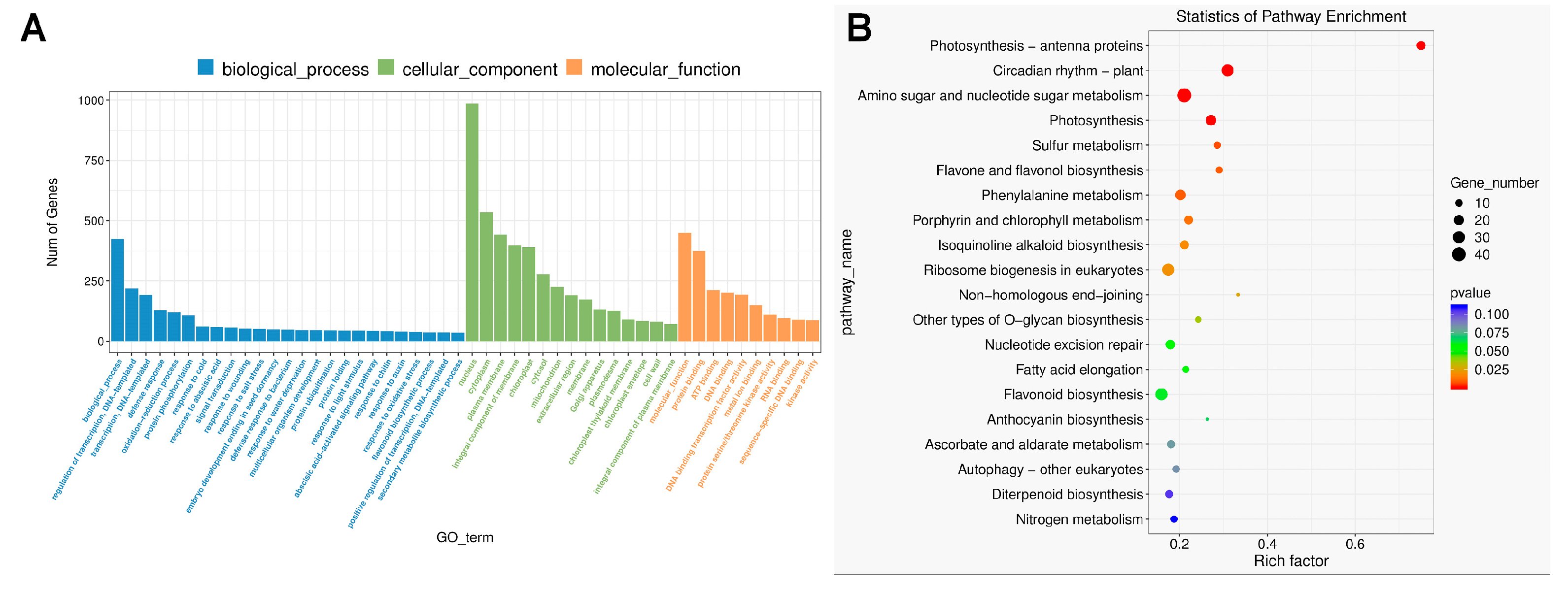
3.1. Transcriptomic Analysis of Two Developmental Stages of Sweet Cheery Fruit
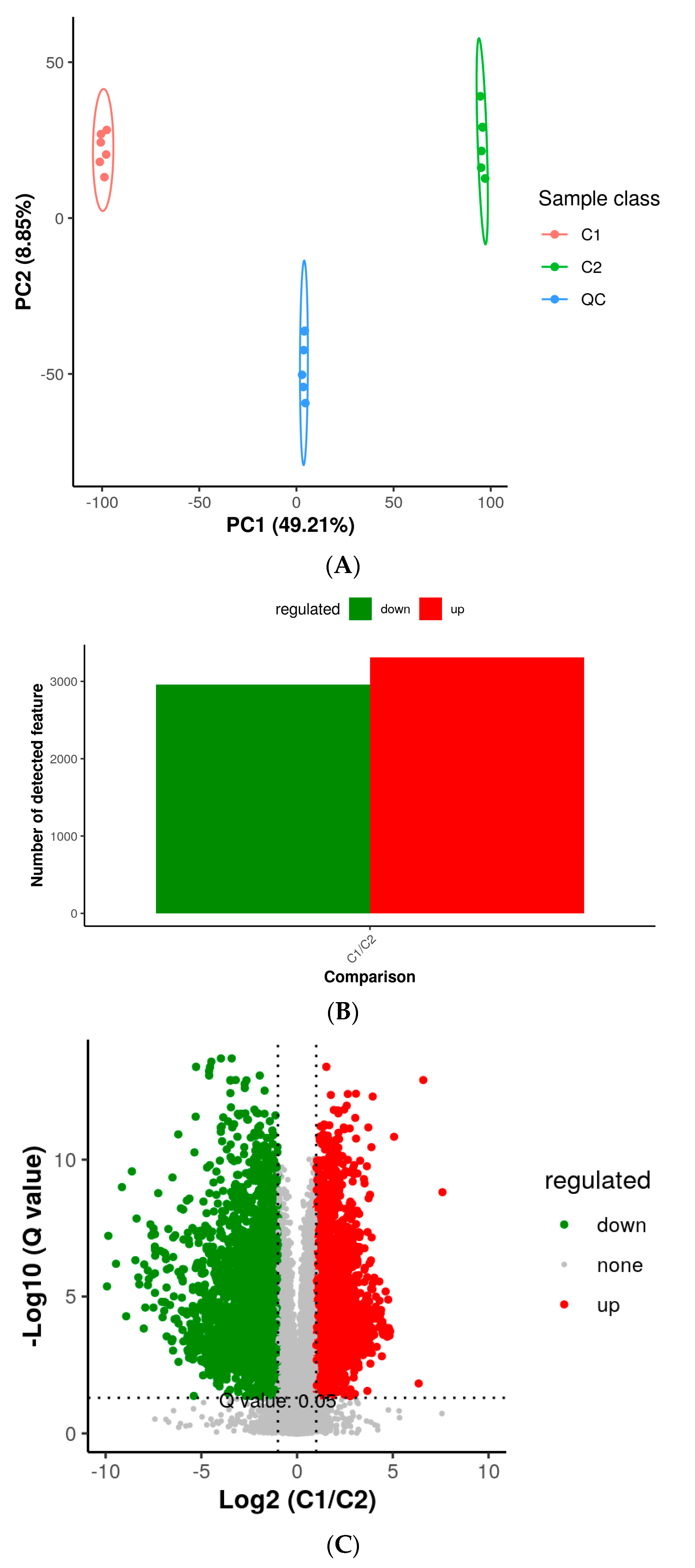
3.2. Metabolic Differences in Two Developmental Stages
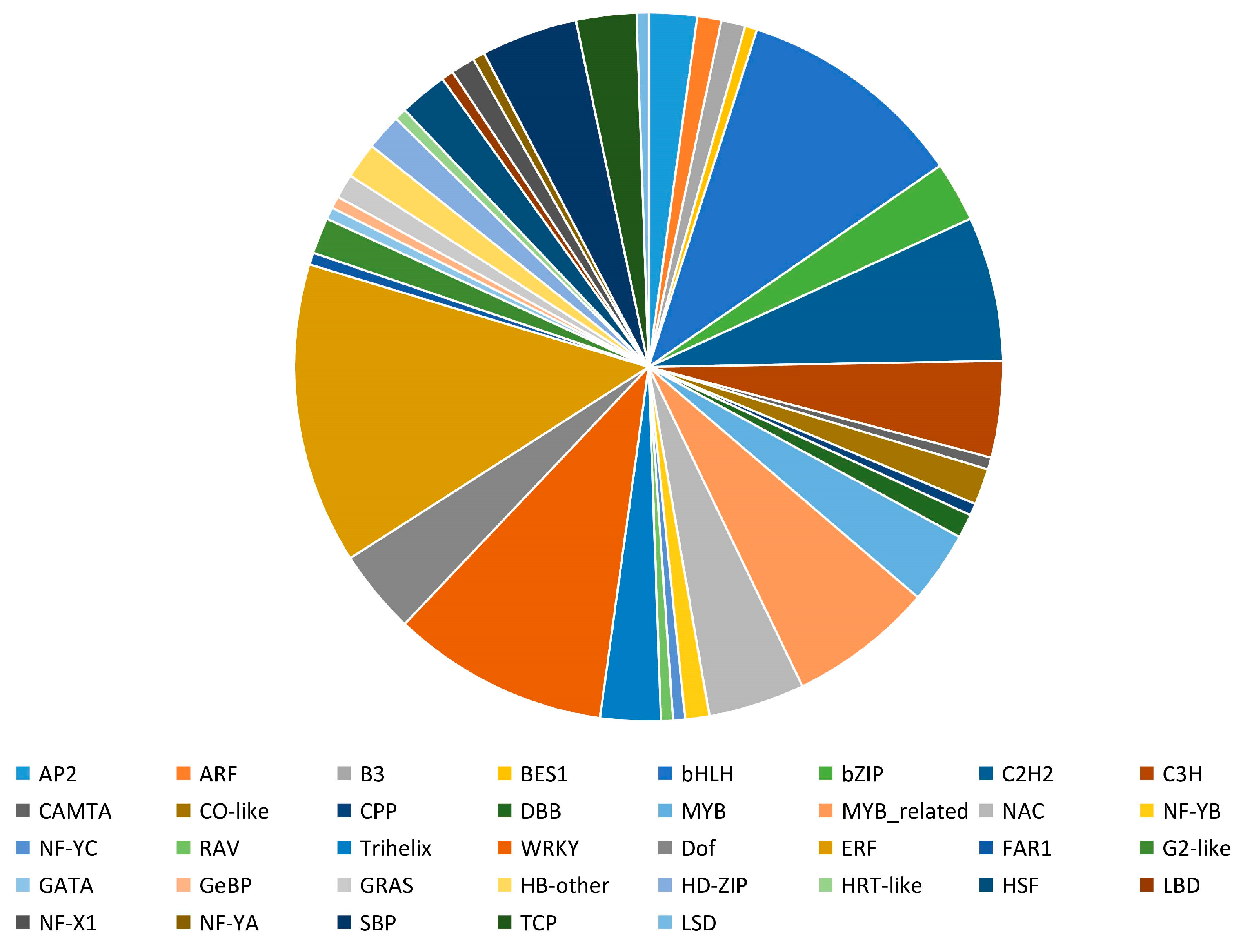
3.3. TFs Are Relevant to Differential Accumulation of Metabolites in “Red Peel” (C2) Sweet Cherries
3.4. Anthocyanin, Flavonoid, and Flavonol Biosynthesis Pathways
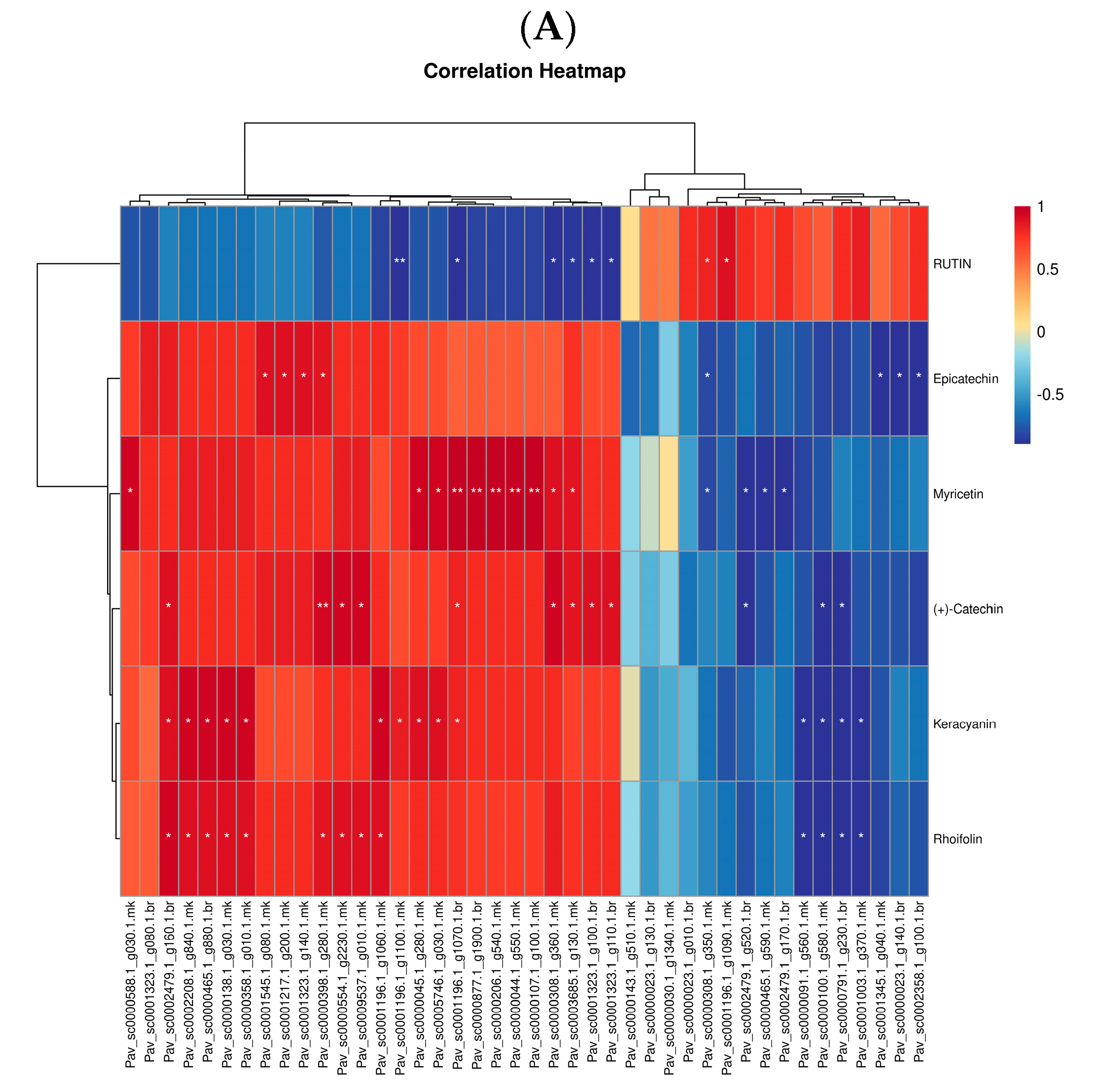
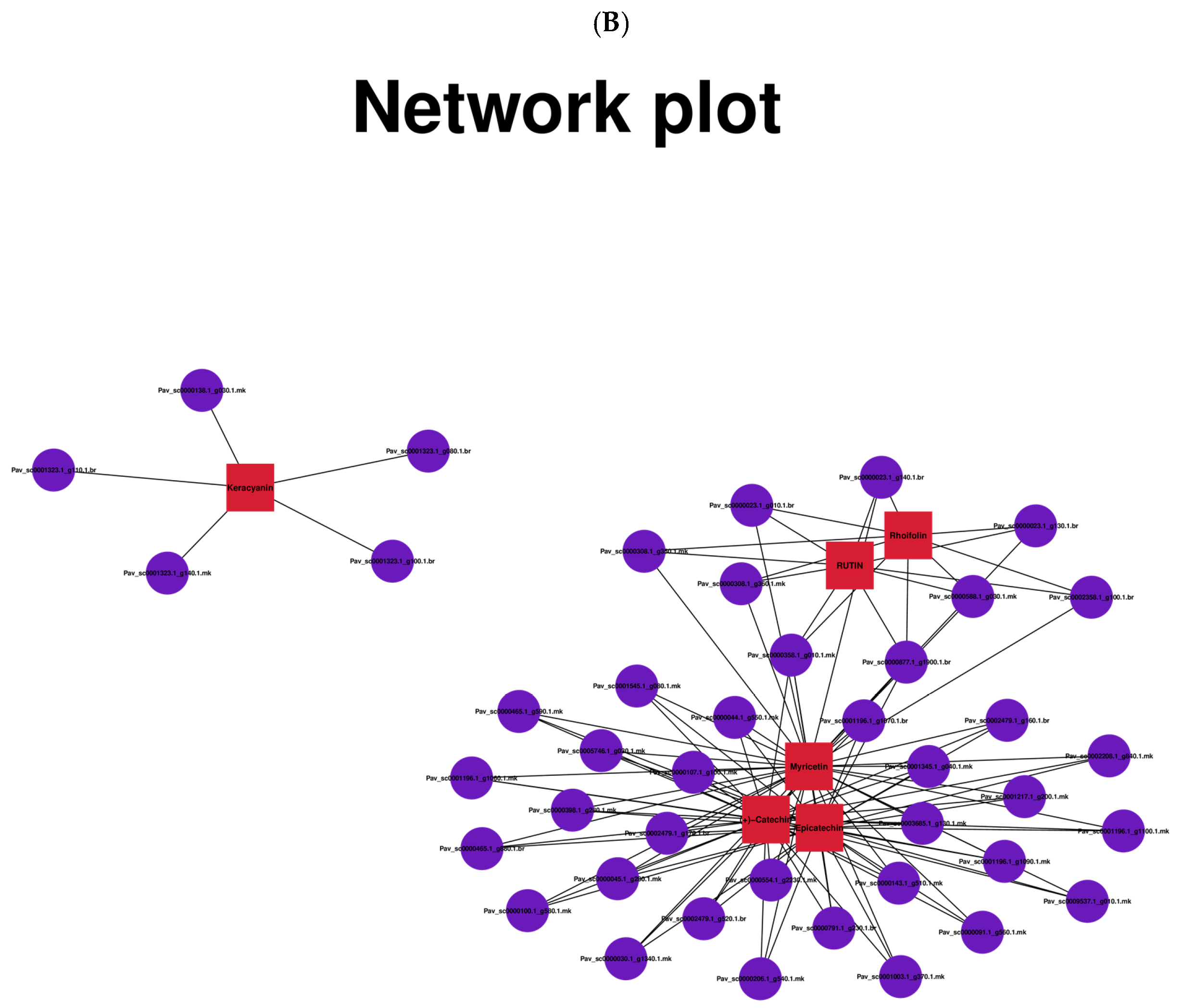
3.5. Correlation Analysis between DEGs and DEMs in Anthocyanin, Flavonoid, Flavone, and Flavanol Biosynthesis Pathways Reveals the Differential Regulatory Network
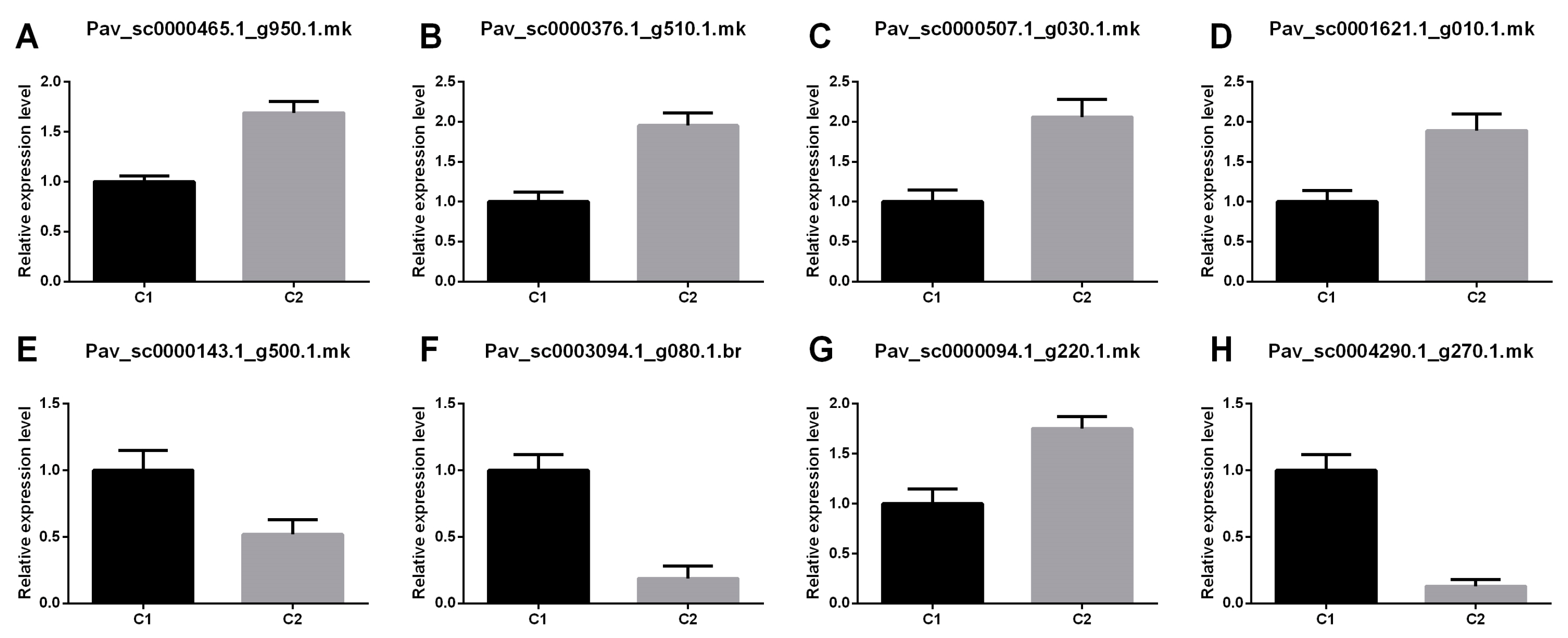
3.6. Verification of the Expression of Genes Relevant to the Route of Flavonoid Biosynthesis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jin, W.; Wang, H.; Li, M.; Wang, J.; Yang, Y.; Zhang, X.; Yan, G.; Zhang, H.; Liu, J.; Zhang, K. The R2R3 MYB transcription factor PavMYB10.1 involves in anthocyanin biosynthesis and determines fruit skin colour in sweet cherry (Prunus avium L.). Plant Biotechnol. J. 2016, 14, 2120–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Chen, X.; Zong, X.; Shu, H.; Gao, D.; Liu, Q. Comparative transcriptome analysis of genes involved in anthocyanin biosynthesis in the red and yellow fruits of sweet cherry (Prunus avium L.). PLoS ONE 2015, 10, e0121164. [Google Scholar] [CrossRef] [PubMed]
- Khoso, M.A.; Hussain, A.; Ritonga, F.N.; Ali, Q.; Channa, M.M.; Alshegaihi, R.M.; Meng, Q.; Ali, M.; Zaman, W.; Brohi, R.D.; et al. WRKY transcription factors (TFs): Molecular switches to regulate drought, temperature, and salinity stresses in plants. Front. Plant Sci. 2022, 13, 1039329. [Google Scholar] [CrossRef]
- Khan, M.; Ali, S.; Manghwar, H.; Saqib, S.; Ullah, F.; Ayaz, A.; Zaman, W. Melatonin Function and Crosstalk with Other Phytohormones under Normal and Stressful Conditions. Genes 2022, 13, 1699. [Google Scholar] [CrossRef]
- Su, L.; Rahat, S.; Ren, N.; Kojima, M.; Takebayashi, Y.; Sakakibara, H.; Wang, M.; Chen, X.; Qi, X. Cytokinin and auxin modulate cucumber parthenocarpy fruit development. Sci. Hortic. 2021, 282, 110026. [Google Scholar] [CrossRef]
- Kong, M.; Sheng, T.; Liang, J.; Ali, Q.; Gu, Q.; Wu, H.; Chen, J.; Liu, J.; Gao, X. Melatonin and Its Homologs Induce Immune Responses via Receptors trP47363-trP13076 in Nicotiana benthamiana. Front. Plant Sci. 2021, 12, 691835. [Google Scholar] [CrossRef]
- Raza, A.; Tabassum, J.; Fakhar, A.Z.; Sharif, R.; Chen, H.; Zhang, C.; Ju, L.; Fotopoulos, V.; Siddique, K.H.M.; Singh, R.K.; et al. Smart reprograming of plants against salinity stress using modern biotechnological tools. Crit. Rev. Biotechnol. 2022, 42, 1–28. [Google Scholar] [CrossRef]
- Sharif, R.; Su, L.; Chen, X.; Qi, X. Hormonal interactions underlying parthenocarpic fruit formation in horticultural crops. Hortic. Res. 2022, 9, uhab024. [Google Scholar] [CrossRef]
- Michailidis, M.; Karagiannis, E.; Tanou, G.; Karamanoli, K.; Lazaridou, A.; Matsi, T.; Molassiotis, A. Metabolomic and physico-chemical approach unravel dynamic regulation of calcium in sweet cherry fruit physiology. Plant Physiol. Biochem. PPB 2017, 116, 68–79. [Google Scholar] [CrossRef]
- Martínez-Esplá, A.; Zapata, P.J.; Valero, D.; García-Viguera, C.; Castillo, S.; Serrano, M. Preharvest application of oxalic acid increased fruit size, bioactive compounds, and antioxidant capacity in sweet cherry cultivars (Prunus avium L.). J. Agric. Food Chem. 2014, 62, 3432–3437. [Google Scholar] [CrossRef]
- Mohotti, S.; Rajendran, S.; Muhammad, T.; Strömstedt, A.A.; Adhikari, A.; Burman, R.; de Silva, E.D.; Göransson, U.; Hettiarachchi, C.M.; Gunasekera, S. Screening for bioactive secondary metabolites in Sri Lankan medicinal plants by microfractionation and targeted isolation of antimicrobial flavonoids from Derris scandens. J. Ethnopharmacol. 2020, 246, 112158. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Li, R.; Ren, L.; Gao, X.; Zhang, Y.; Ma, Z.; Ma, D.; Luo, Y. A comparative metabolomics study of flavonoids in sweet potato with different flesh colors (Ipomoea batatas (L.) Lam). Food Chem. 2018, 260, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Casili, G.; Lanza, M.; Campolo, M.; Messina, S.; Scuderi, S.; Ardizzone, A.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Therapeutic potential of flavonoids in the treatment of chronic venous insufficiency. Vasc. Pharmacol. 2021, 137, 106825. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Brugliera, F.; Chandler, S. Recent progress of flower colour modification by biotechnology. Int. J. Mol. Sci. 2009, 10, 5350–5369. [Google Scholar] [CrossRef]
- Pelletier, M.K.; Murrell, J.R.; Shirley, B.W. Characterization of flavonol synthase and leucoanthocyanidin dioxygenase genes in Arabidopsis. Further evidence for differential regulation of “early” and “late” genes. Plant Physiol. 1997, 113, 1437–1445. [Google Scholar] [CrossRef] [Green Version]
- Li, X.W.; Li, J.W.; Zhai, Y.; Zhao, Y.; Zhao, X.; Zhang, H.J.; Su, L.T.; Wang, Y.; Wang, Q.Y. A R2R3-MYB transcription factor, GmMYB12B2, affects the expression levels of flavonoid biosynthesis genes encoding key enzymes in transgenic Arabidopsis plants. Gene 2013, 532, 72–79. [Google Scholar] [CrossRef]
- Wei, J.; Yang, J.; Jiang, W.; Pang, Y. Stacking triple genes increased proanthocyanidins level in Arabidopsis thaliana. PLoS ONE 2020, 15, e0234799. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, W.; Liu, J.; Liu, H.; Lv, Z.; Zhang, C.; Chen, D.; Jiao, Z. Postharvest UV-C irradiation increased the flavonoids and anthocyanins accumulation, phenylpropanoid pathway gene expression, and antioxidant activity in sweet cherries (Prunus avium L.). Postharvest Biol. Technol. 2021, 175, 111490. [Google Scholar] [CrossRef]
- Qi, X.; Liu, C.; Song, L.; Li, M. PaMADS7, a MADS-box transcription factor, regulates sweet cherry fruit ripening and softening. Plant Sci. 2020, 301, 110634. [Google Scholar] [CrossRef]
- Dong, Y.; Qi, X.; Liu, C.; Song, L.; Ming, L. A sweet cherry AGAMOUS-LIKE transcription factor PavAGL15 affects fruit size by directly repressing the PavCYP78A9 expression. Sci. Hortic. 2022, 297, 110947. [Google Scholar] [CrossRef]
- Wang, J.; Sun, W.; Wang, L.; Liu, X.; Xu, Y.; Sabir, I.A.; Jiu, S.; Wang, S.; Zhang, C. FRUITFULL is involved in double fruit formation at high temperature in sweet cherry. Environ. Exp. Bot. 2022, 201, 104986. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Yang, W.; Li, J.; Tang, W.; Gong, R. Transcriptomic and Metabolomic Analysis of Quality Changes during Sweet Cherry Fruit Development and Mining of Related Genes. Int. J. Mol. Sci. 2022, 23, 7402. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Kusano, M.; Redestig, H. Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 2009, 13, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, N.; Maldonado, J.; Ponce, C.; Arellano, M.; Time, A.; Multari, S.; Martens, S.; Carrera, E.; Donoso, J.M.; Sagredo, B.; et al. RNAseq reveals different transcriptomic responses to GA3 in early and midseason varieties before ripening initiation in sweet cherry fruits. Sci. Rep. 2021, 11, 13075. [Google Scholar] [CrossRef]
- Li, Y.K.; Fang, J.B.; Qi, X.J.; Lin, M.M.; Zhong, Y.P.; Sun, L.M.; Cui, W. Combined analysis of the fruit metabolome and transcriptome reveals candidate genes involved in flavonoid biosynthesis in Actinidia arguta. Int. J. Mol. Sci. 2018, 19, 1471. [Google Scholar] [CrossRef] [Green Version]
- Kleessen, S.; Irgang, S.; Klie, S.; Giavalisco, P.; Nikoloski, Z. Integration of transcriptomics and metabolomics data specifies Chlamydomonas’ metabolic response to rapamycin tratment. Plant J. 2015, 81, 822–835. [Google Scholar] [CrossRef]
- Glaubitz, U.; Li, X.; Schaedel, S.; Erban, A.; Sulpice, R.; Kopka, J.; Hincha, D.K.; Zuther, E. Integrated analysis of rice transcriptomic and metabolomic responses to elevated night temperatures identifies sensitivity- and tolerance-related profiles: Integrated profiling analysis of rice under HNT. Plant Cell Environ. 2017, 40, 121–137. [Google Scholar] [CrossRef]
- Hunter, D.A.; Napier, N.J.; Erridge, Z.A.; Saei, A.; Chen, R.K.Y.; McKenzie, M.J.; O’Donoghue, E.M.; Hunt, M.; Favre, L.; Lill, R.E.; et al. Transcriptome Responses of Ripe Cherry Tomato Fruit Exposed to Chilling and Rewarming Identify Reversible and Irreversible Gene Expression Changes. Front. Plant Sci. 2021, 12, 685416. [Google Scholar] [CrossRef]
- Cho, K.; Cho, K.S.; Sohn, H.B.; Ha, I.J.; Hong, S.Y.; Lee, H.; Kim, Y.M.; Nam, M.H. Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J. Exp. Bot. 2016, 67, 1519. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wu, J.; Li, S.S.; Zhang, H.J.; Feng, C.Y.; Yin, D.D.; Wu, R.Y.; Wang, L.S. Transcriptome sequencing and metabolite analysis for revealing the blue flower formation in waterlily. BMC Genom. 2016, 17, 897. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.F.; Han, Z.X.; Feng, L.; Gao, L.P.; Gao, M.J.; Gruber, M.Y.; Zhang, Z.L.; Xia, T.; Wan, X.C.; Wei, S. Metabolic flux redirection and transcriptomic reprogramming in the albino tea cultivar ‘Yu-Jin-Xiang’ with an emphasis on catechin production. Sci. Rep. 2017, 7, 45062. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.T.; Wang, S.C.; Huang, Z.J.; Zhang, S.B.; Liao, Q.G.; Zhang, C.Z.; Lin, T.; Qin, M.; Peng, M.; Yang, C.K.; et al. Rewiring of the fruit metabolome in tomato breeding. Cell 2018, 172, 249–261.e12. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.U.; Nupur, J.A.; Khalid, M.H.B.; Din, A.M.U.; Shafiq, M.; Alshegaihi, R.M.; Ali, Q.; Ali, Q.; Kamran, Z.; Manzoor, M.; et al. Genome-Wide Identification and In Silico Analysis of ZF-HD Transcription Factor Genes in Zea mays L. Genes 2022, 13, 2112. [Google Scholar] [CrossRef]
- Poux, S.; Arighi, C.N.; Magrane, M.; Bateman, A.; Wei, C.H.; Lu, Z.; Boutet, E.; Bye, A.J.H.; Famiglietti, M.L.; Roechert, B.; et al. On expert curation and scalability: UniProtKB/Swiss-Prot as a case study. Bioinformatics 2017, 33, 3454–3460. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-discovery approach for sample matching of a nerve-agent precursor using liquid chromatography-mass spectrometry, XCMS, and chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- Wang, M.; Avula, B.; Wang, Y.H.; Zhao, J.; Avonto, C.; Parcher, J.F.; Raman, V.; Zweigenbaum, J.A.; Wylie, P.L.; Khan, I.A. An integrated approach utilising chemometrics and GC/MS for classification of chamomile flowers, essential oils and commercial products. Food Chem. 2014, 152, 391–398. [Google Scholar] [CrossRef]
- Guo, H.H.; Guo, H.X.; Zhang, L.; Tang, Z.M.; Yu, X.M.; Wu, J.F.; Zeng, F.C. Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int. J. Mol. Sci. 2019, 20, 2070. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.; Beale, M.; Fiehn, O.; Hardy, N.; Sumner, L.; Bino, R. Plant metabolomics: The missing link in functional genomics strategies. Plant Cell 2002, 14, 1437–1440. [Google Scholar] [CrossRef] [Green Version]
- Jalal, A.; Ali, Q.; Manghwar, H.; Zhu, D. Identification, Phylogeny, Divergence, Structure, and Expression Analysis of A20/AN1 Zinc Finger Domain Containing Stress-Associated Proteins (SAPs) Genes in Jatropha curcas L. Genes 2022, 13, 1766. [Google Scholar] [CrossRef]
- Mehrtens, F.; Kranz, H.; Bednarek, P.; Weisshaar, B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138, 1083–1096. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Osbourn, A.; Ma, P. MYB Transcription Factors as Regulators of Phenylpropanoid Metabolism in Plants. Mol. Plant 2015, 8, 689–708. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yu, Q.; Shen, W.; El Mohtar, C.A.; Zhao, X.; Gmitter, F.G., Jr. Functional study of CHS gene family members in citrus revealed a novel CHS gene affecting the production of flavonoids. BMC Plant Biol. 2018, 18, 189. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, G.; Chen, J.; Liu, X.; Lu, X.; Chen, H.; Tian, Y. Integrated Metabolome and Transcriptome Analysis Unveils Novel Pathway Involved in the Formation of Yellow Peel in Cucumber. Int. J. Mol. Sci. 2021, 22, 1494. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pu, H.; Shan, S.; Zhang, P.; Li, J.; Song, H.; Xu, X. Melatonin enhanced chilling tolerance and alleviated peel browning of banana fruit under low temperature storage. Postharvest Biol. Technol. 2021, 179, 111571. [Google Scholar] [CrossRef]
- Sivankalyani, V.; Feygenberg, O.; Diskin, S.; Wright, B.; Alkan, N. Increased anthocyanin and flavonoids in mango fruit peel are associated with cold and pathogen resistance. Postharvest Biol. Technol. 2016, 111, 132–139. [Google Scholar] [CrossRef]
- Naeem, M.; Shahzad, K.; Saqib, S.; Shahzad, A.; Nasrullah; Younas, M.; Afridi, M.I. The Solanum melongena COP1LIKE manipulates fruit ripening and flowering time in tomato (Solanum lycopersicum). Plant Growth Regul. 2022, 96, 369–382. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Huang, R.; Liu, L.; Li, Y.; Wang, W.; Xu, Q.; Yu, Y.; Zhou, T. CsUGT78A15 catalyzes the anthocyanidin 3-O-galactoside biosynthesis in tea plants. Plant Physiol. Biochem. PPB 2021, 166, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Ranganath, K.G.; Shivashankara, K.S.; Roy, T.K.; Dinesh, M.R.; Geetha, G.A.; Pavithra, K.C.; Ravishankar, K.V. Profiling of anthocyanins and carotenoids in fruit peel of different colored mango cultivars. J. Food Sci. Technol. 2018, 55, 4566–4577. [Google Scholar] [CrossRef]
- Liu, Y.; Tikunov, Y.; Schouten, R.E.; Marcelis, L.F.M.; Visser, R.G.F.; Bovy, A. Anthocyanin Biosynthesis and Degradation Mechanisms in Solanaceous Vegetables: A Review. Front. Chem. 2018, 6, 52. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Liu, Z.; Zhao, Z.; Zhao, J.; Wang, Z.; Zhou, G.; Liu, P.; Liu, M. Transcriptome and metabolome profiling unveil the mechanisms of Ziziphus jujuba Mill. peel coloration. Food Chem. 2020, 312, 125903. [Google Scholar] [CrossRef]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. Cell Mol. Biol. 2011, 66, 94–116. [Google Scholar] [CrossRef]
- Wang, X.-Y.; Tian, L.; Feng, S.-J.; Wei, A.-Z. Identifying potential flavonoid biosynthesis regulator in Zanthoxylum bungeanum Maxim. by genome-wide characterization of the MYB transcription factor gene family. J. Integr. Agric. 2022, 21, 1997–2018. [Google Scholar] [CrossRef]
- Zhu, L.; Guan, Y.; Zhang, Z.; Song, A.; Chen, S.; Jiang, J.; Chen, F. CmMYB8 encodes an R2R3 MYB transcription factor which represses lignin and flavonoid synthesis in chrysanthemum. Plant Physiol. Biochem. 2020, 149, 217–224. [Google Scholar] [CrossRef]
- Zhu, J.-H.; Xia, D.-N.; Xu, J.; Guo, D.; Li, H.-L.; Wang, Y.; Mei, W.-L.; Peng, S.-Q. Identification of the bHLH gene family in Dracaena cambodiana reveals candidate genes involved in flavonoid biosynthesis. Ind. Crops Prod. 2020, 150, 112407. [Google Scholar] [CrossRef]
- Arlotta, C.; Puglia, G.D.; Genovese, C.; Toscano, V.; Karlova, R.; Beekwilder, J.; De Vos, R.C.H.; Raccuia, S.A. MYB5-like and bHLH influence flavonoid composition in pomegranate. Plant Sci. 2020, 298, 110563. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Z.; Zeng, Q.; Wang, S.; Luo, Y.; Huang, Y.; Xin, Y.; He, N. Abnormal expression of bHLH3 disrupts a flavonoid homeostasis network, causing differences in pigment composition among mulberry fruits. Hortic. Res. 2020, 7, 83. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.; Misra, P.; Trivedi, P.K.; Pandey, A. Molecular components associated with the regulation of flavonoid biosynthesis. Plant Sci. 2022, 317, 111196. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Kopka, J.; Dormann, P.; Altmann, T.; Trethewey, R.N.; Willmitzer, L. Metabolite profiling for plant functional genomics. Nat. Biotechnol. 2000, 18, 1157–1161. [Google Scholar] [CrossRef]
- Liu, Y.; Lv, J.; Liu, Z.; Wang, J.; Yang, B.; Chen, W.; Ou, L.; Dai, X.; Zhang, Z.; Zou, X. Integrative analysis of metabolome and transcriptome reveals the mechanism of color formation in pepper fruit (Capsicum annuum L.). Food Chem. 2020, 306, 125629. [Google Scholar] [CrossRef]
- Appelhagen, I.; Lu, G.H.; Huep, G.; Schmelzer, E.; Weisshaar, B.; Sagasser, M. TRANSPARENT TESTA1 interacts with R2R3-MYB factors and affects early and late steps of flavonoid biosynthesis in the endothelium of Arabidopsis thaliana seeds. Plant J. Cell Mol. Biol. 2011, 67, 406–419. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway_id | Pathway_Name | Gene_ID | Description | Regulation |
|---|---|---|---|---|
| ko00942 | Anthocyanin biosynthesis | Pav_sc0000138.1_g030.1.mk | anthocyanidin 3-O-glucosyltransferase 2-like [Prunus avium] | down |
| ko00942 | Anthocyanin biosynthesis | Pav_sc0001323.1_g080.1.br | UDP-glycosyltransferase 88F5-like [Prunus avium] | down |
| ko00942 | Anthocyanin biosynthesis | Pav_sc0001323.1_g100.1.br | UDP-glycosyltransferase 88F5-like [Prunus avium] | down |
| ko00942 | Anthocyanin biosynthesis | Pav_sc0001323.1_g110.1.br | UDP-glycosyltransferase 88F5-like [Prunus avium] | down |
| ko00942 | Anthocyanin biosynthesis | Pav_sc0001323.1_g140.1.mk | LOW QUALITY PROTEIN: UDP-glycosyltransferase 88F5-like [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000030.1_g1340.1.mk | flavonol synthase/flavanone 3-hydroxylase-like [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000044.1_g550.1.mk | naringenin,2-oxoglutarate 3-dioxygenase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000045.1_g280.1.mk | chalcone synthase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000091.1_g560.1.mk | ELMO domain-containing protein A [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000100.1_g580.1.mk | protein ECERIFERUM 26-like [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000107.1_g100.1.mk | leucoanthocyanidin dioxygenase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000143.1_g510.1.mk | probable 2-oxoglutarate-dependent dioxygenase At5g05600 [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000206.1_g540.1.mk | trans-cinnamate 4-monooxygenase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000358.1_g010.1.mk | flavonoid-3′-hydroxylase, partial [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000398.1_g280.1.mk | putative anthocyanidin reductase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000465.1_g590.1.mk | codeine O-demethylase-like [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000465.1_g880.1.br | codeine O-demethylase-like [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000554.1_g2230.1.mk | probable LRR receptor-like serine/threonine-protein kinase MEE39 [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000791.1_g230.1.br | BAHD acyltransferase At5g47980-like [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0000877.1_g1900.1.br | flavonoid 3′-monooxygenase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001003.1_g370.1.mk | vacuolar-sorting receptor 7 isoform X2 [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001196.1_g1060.1.mk | cytochrome P450 98A2-like isoform X1 [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001196.1_g1070.1.br | cytochrome P450 98A2-like isoform X1 [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001196.1_g1090.1.mk | cytochrome P450 98A2-like [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001196.1_g1100.1.mk | cytochrome P450 98A2 [Prunus persica] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001217.1_g200.1.mk | protein DMR6-LIKE OXYGENASE 2-like [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001345.1_g040.1.mk | probable 2-oxoglutarate-dependent dioxygenase At3g111800 [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0001545.1_g080.1.mk | hypothetical protein PRUPE_4G029700 [Prunus persica] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0002208.1_g840.1.mk | bifunctional dihydroflavonol 4-reductase/flavanone 4-reductase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0002479.1_g160.1.br | vinorine synthase-like [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0002479.1_g170.1.br | uncharacterized protein Pyn_04376 [Prunus yedoensis var. nudiflora] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0002479.1_g520.1.br | vinorine synthase-like [Prunus avium] | up |
| ko00941 | Flavonoid biosynthesis | Pav_sc0003685.1_g130.1.mk | leucoanthocyanidin reductase [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0005746.1_g030.1.mk | probable chalcone—flavonone isomerase 3 [Prunus avium] | down |
| ko00941 | Flavonoid biosynthesis | Pav_sc0009537.1_g010.1.mk | chalcone synthase [Prunus yedoensis var. nudiflora] | down |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000023.1_g010.1.br | phenolic glucoside malonyltransferase 1-like [Prunus avium] | up |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000023.1_g130.1.br | phenolic glucoside malonyltransferase 1-like [Prunus avium] | up |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000023.1_g140.1.br | phenolic glucoside malonyltransferase 1-like [Prunus avium] | up |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000308.1_g350.1.mk | flavonoid 3-O-glucosyltransferase-like [Prunus avium] | up |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000308.1_g360.1.mk | flavonoid 3-O-glucosyltransferase-like [Prunus avium] | down |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000358.1_g010.1.mk | flavonoid-3′-hydroxylase, partial [Prunus avium] | down |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000588.1_g030.1.mk | UDP-glycosyltransferase 89A2-like [Prunus avium] | down |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0000877.1_g1900.1.br | flavonoid 3′-monooxygenase [Prunus avium] | down |
| ko00944 | Flavone and flavonol biosynthesis | Pav_sc0002358.1_g100.1.br | phenolic glucoside malonyltransferase 1-like [Prunus avium] | up |
| Pathway ID | Regulated | MS2 Metabolite | MS2 Superclass | MS2 Class | MS2 KEGG |
|---|---|---|---|---|---|
| map00941 | down | (+)-Catechin | Phenylpropanoids and polyketides | Flavonoids | C06562 |
| map00941 | down | Epicatechin | Phenylpropanoids and polyketides | Flavonoids | C09727 |
| map00941 | down | Myricetin | Phenylpropanoids and polyketides | Flavonoids | C10107 |
| map00944 | down | Rhoifolin | Phenylpropanoids and polyketides | Flavonoids | C12627 |
| map00944 | up | RUTIN | Phenylpropanoids and polyketides | Flavonoids | C05625 |
| map00944 | down | Myricetin | Phenylpropanoids and polyketides | Flavonoids | C10107 |
| map00942 | down | Keracyanin | Phenylpropanoids and polyketides | Flavonoids | C08620 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, B.; Tian, Y. Combined Study of Transcriptome and Metabolome Reveals Involvement of Metabolites and Candidate Genes in Flavonoid Biosynthesis in Prunus avium L. Horticulturae 2023, 9, 463. https://doi.org/10.3390/horticulturae9040463
Fu B, Tian Y. Combined Study of Transcriptome and Metabolome Reveals Involvement of Metabolites and Candidate Genes in Flavonoid Biosynthesis in Prunus avium L. Horticulturae. 2023; 9(4):463. https://doi.org/10.3390/horticulturae9040463
Chicago/Turabian StyleFu, Baochun, and Yongqiang Tian. 2023. "Combined Study of Transcriptome and Metabolome Reveals Involvement of Metabolites and Candidate Genes in Flavonoid Biosynthesis in Prunus avium L." Horticulturae 9, no. 4: 463. https://doi.org/10.3390/horticulturae9040463