
Identification and Comparative Analysis of Genes and MicroRNAs Involved in the Floral Transition of the Xinjiang Early-Flowering Walnut (Juglans regia L.)

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
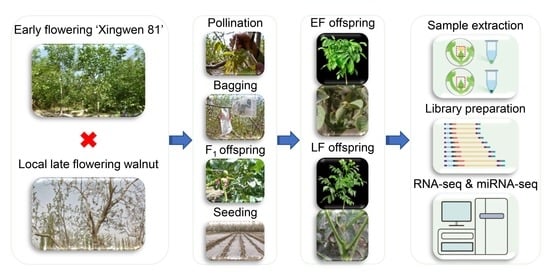
2.1. Plant Materials and Sample Collection
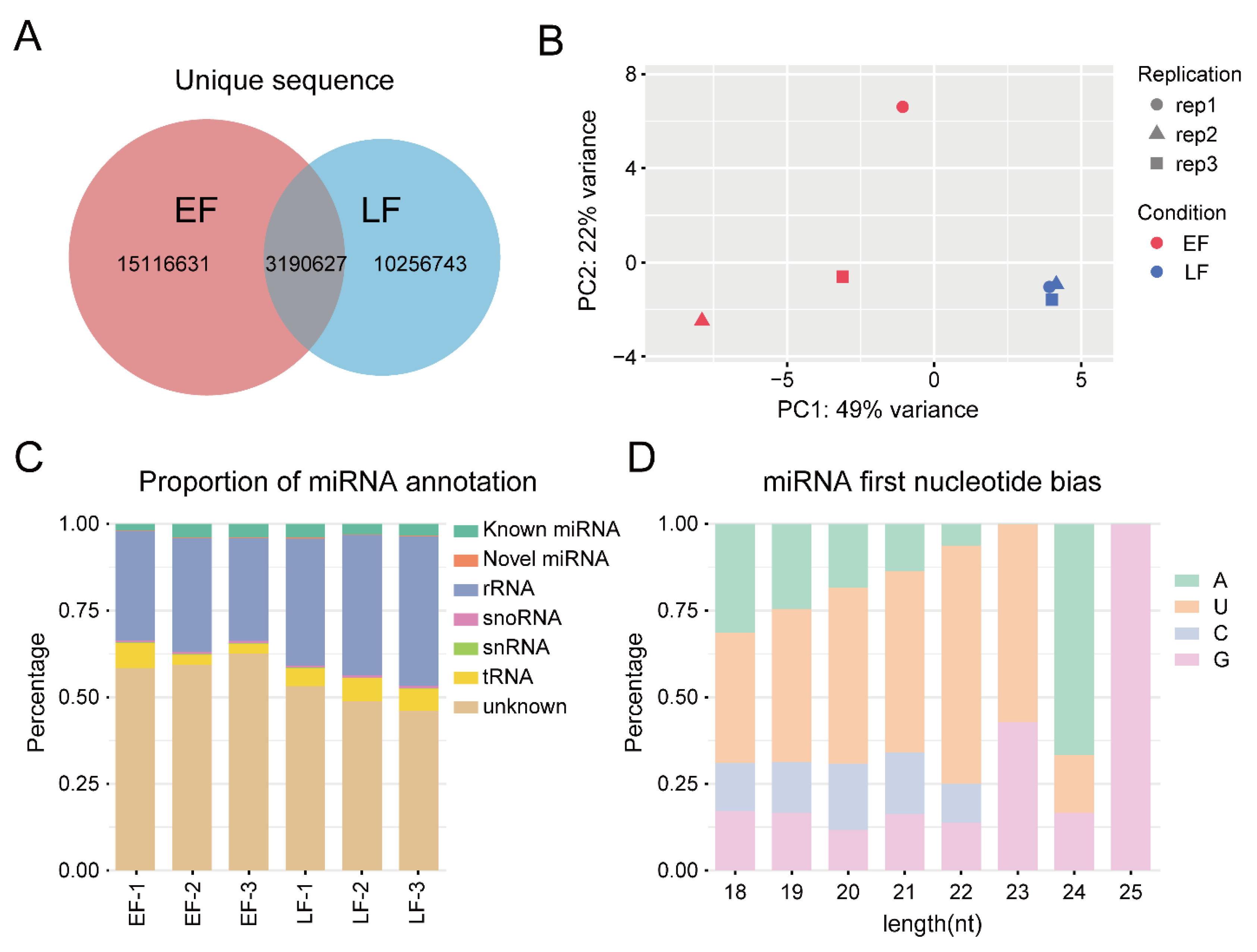
2.2. RNA Extraction, Library Preparation, RNA Sequencing, and microRNA Sequencing
2.3. RNA-Seq and miRNA-Seq Data Analysis
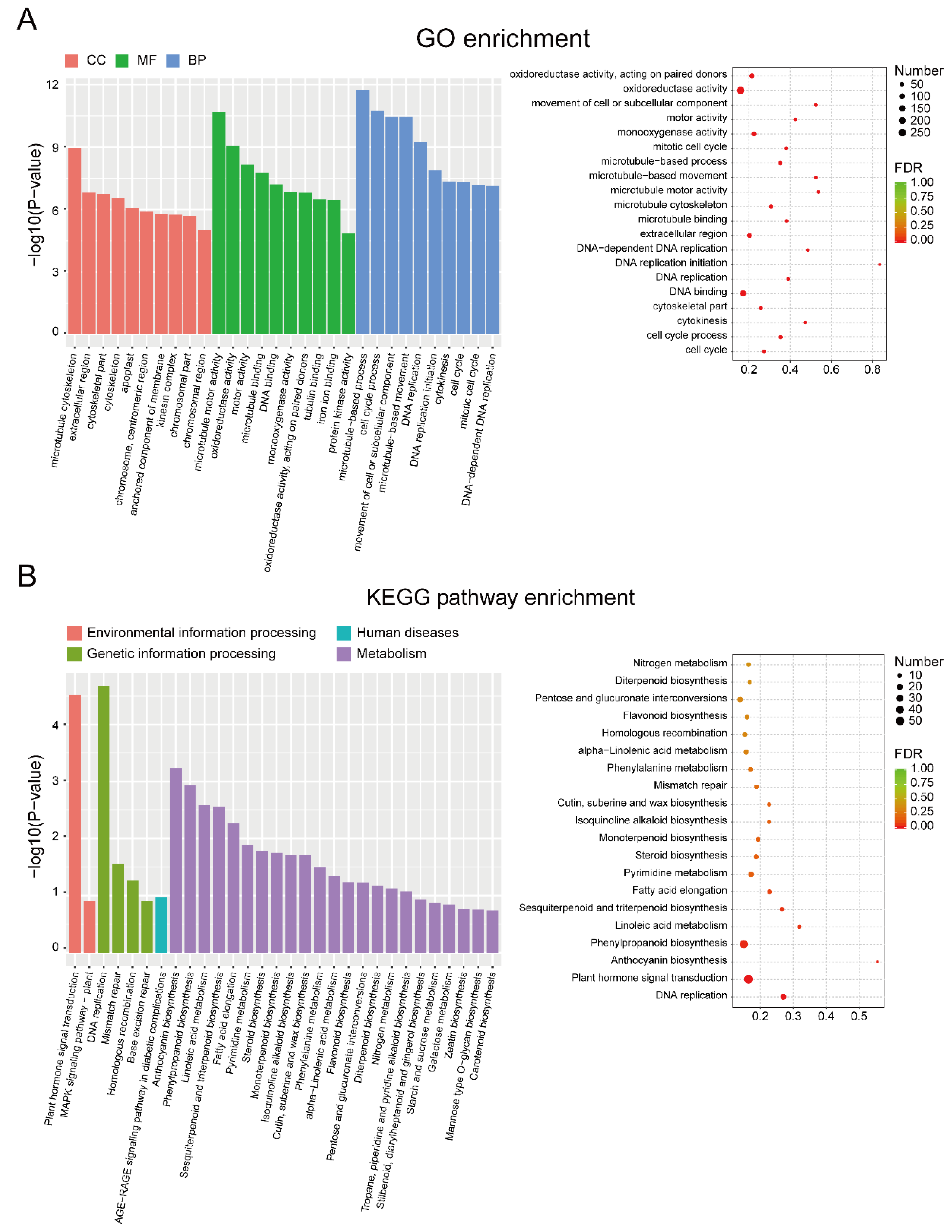
2.4. Functional Enrichment Analysis
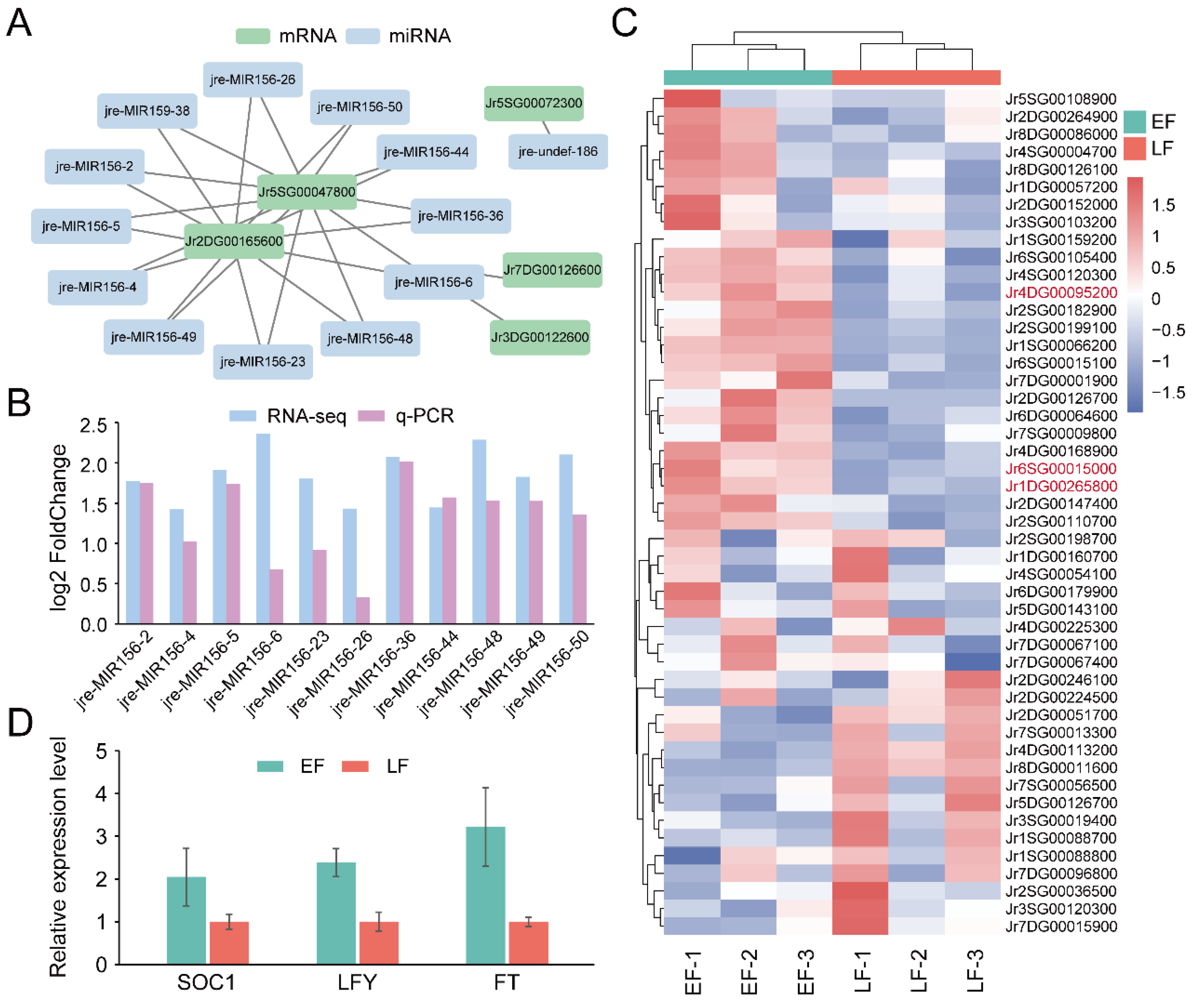
2.5. miRNA Target Prediction
2.6. Quantitative Real-Time PCR Validation
3. Results
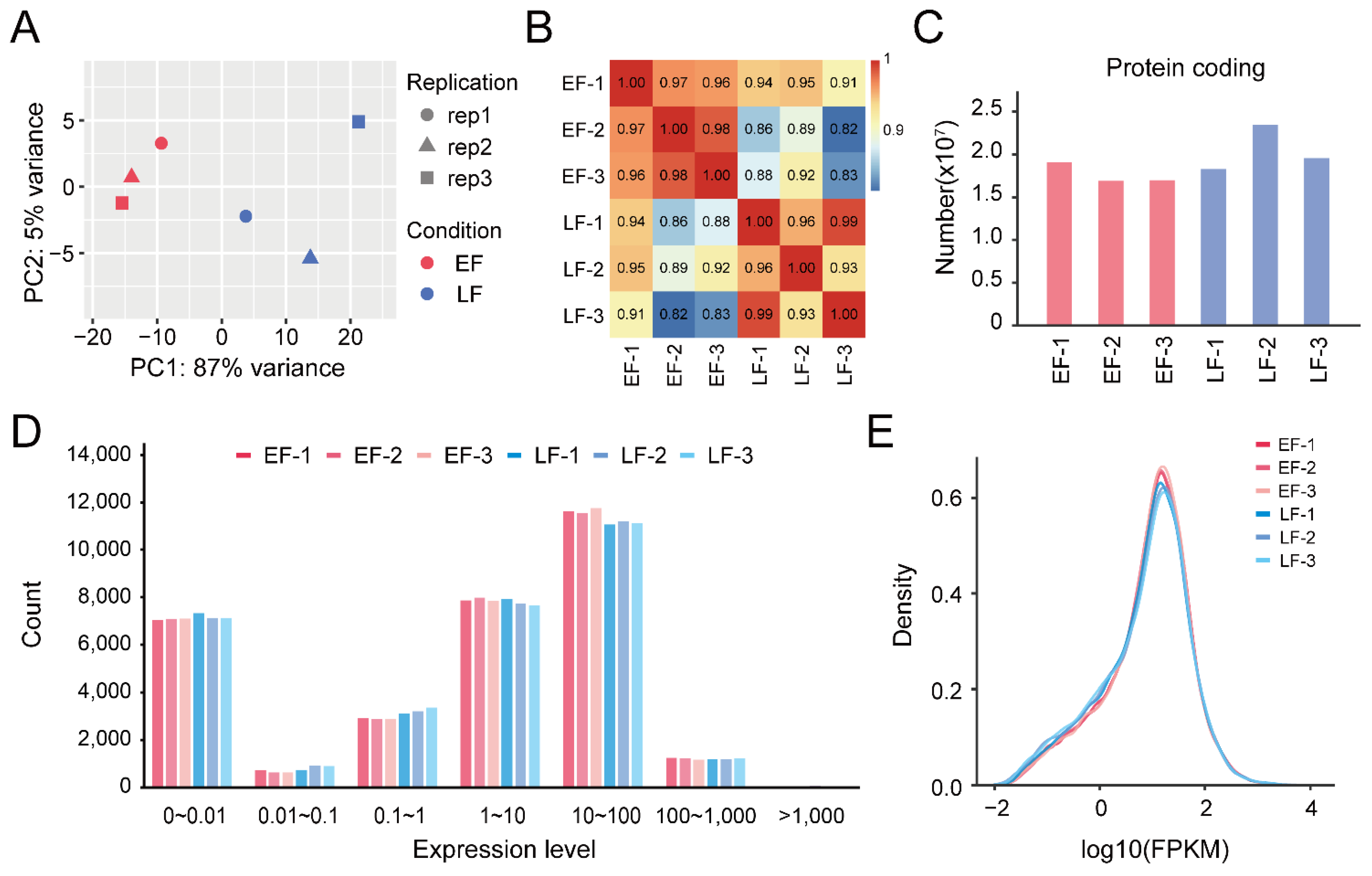
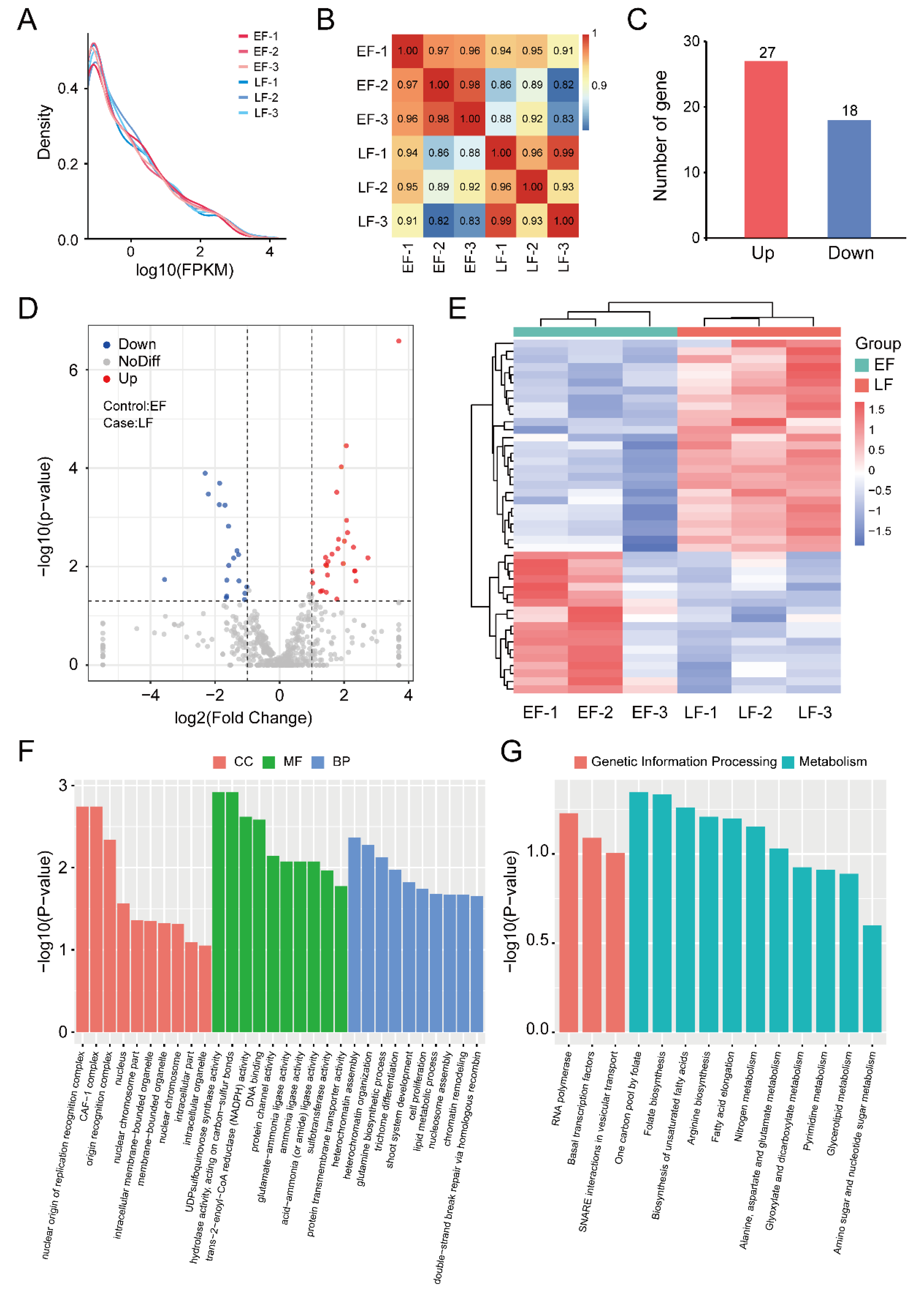
3.1. RNA Sequencing and Transcription Analysis
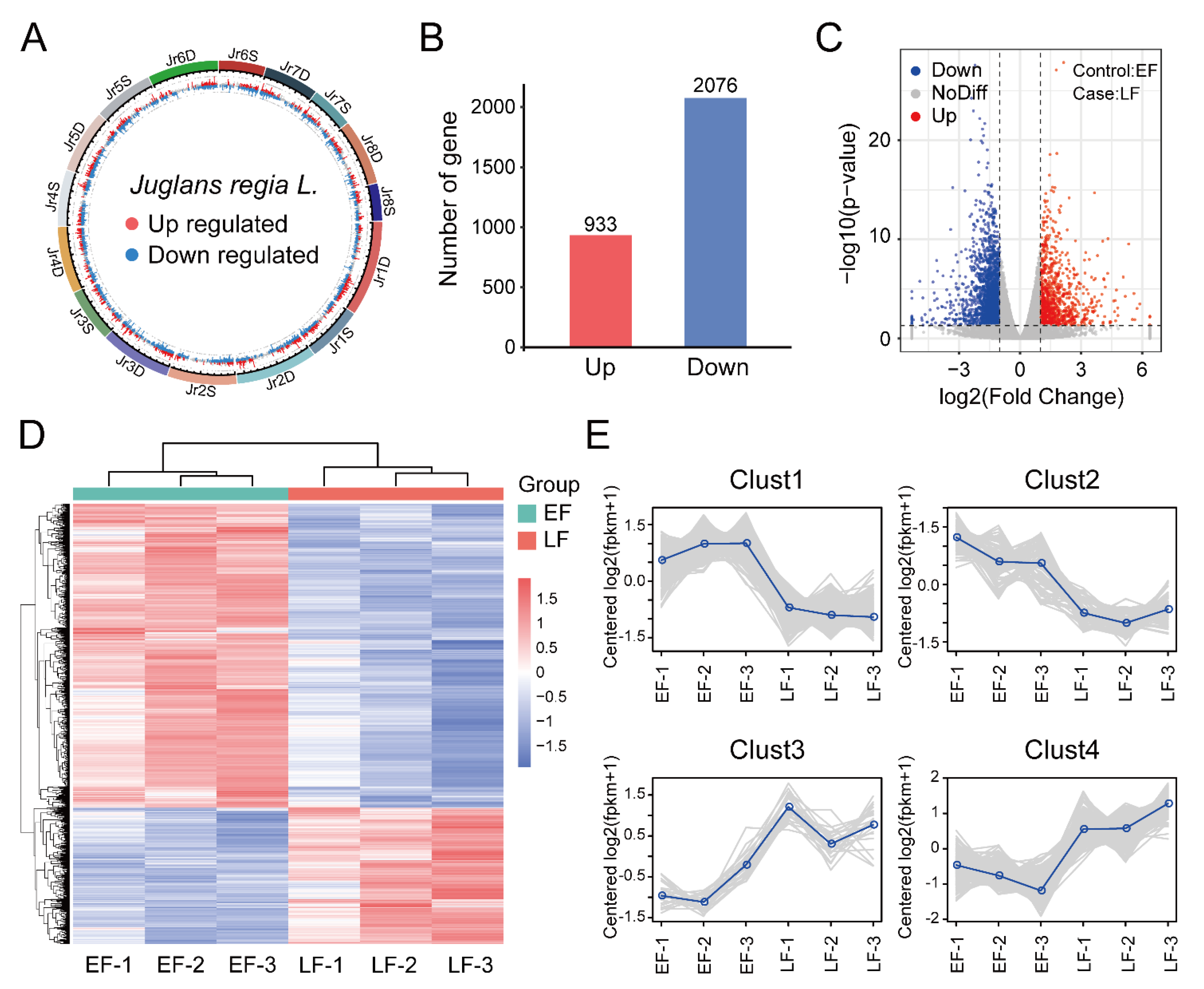
3.2. Identification and Enrichment Analysis of DEGs between EF and LF Walnut Varieties
3.3. miRNA Sequencing and Data Analysis
3.4. Identification of Genes and miRNAs Associated with Flowering Time
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, Y. Chromatin regulation of flowering. Trends Plant Sci. 2012, 17, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Conti, L. Hormonal control of the floral transition: Can one catch them all? Dev. Biol. 2017, 430, 288–301. [Google Scholar] [CrossRef] [PubMed]
- He, Y. Control of the transition to flowering by chromatin modifications. Mol. Plant 2009, 2, 554–564. [Google Scholar] [CrossRef]
- Bouché, F.; Lobet, G.; Tocquin, P.; Périlleux, C. FLOR-ID: An interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016, 44, D1167–D1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Li, X.; Wang, Y.; Guo, W.; Chachar, S.; Riaz, A.; Geng, Y.; Gu, X.; Yang, L. PRMT6 physically associates with nuclear factor Y to regulate photoperiodic flowering in Arabidopsis. aBIOTECH 2021, 2, 403–414. [Google Scholar] [CrossRef]
- Gu, X.; Wang, Y.; He, Y. Photoperiodic regulation of flowering time through periodic histone deacetylation of the florigen gene FT. PLoS Biol. 2013, 11, e1001649. [Google Scholar] [CrossRef]
- Helliwell, C.A.; Wood, C.C.; Robertson, M.; James Peacock, W.; Dennis, E.S. The Arabidopsis FLC protein interacts directly in vivo with SOC1 and FT chromatin and is part of a high-molecular-weight protein complex. Plant J. 2006, 46, 183–192. [Google Scholar] [CrossRef]
- Corbesier, L.; Vincent, C.; Jang, S.; Fornara, F.; Fan, Q.; Searle, I.; Giakountis, A.; Farrona, S.; Gissot, L.; Turnbull, C.; et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 2007, 316, 1030–1033. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Warthmann, N.; Küttner, F.; Schmid, M. Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Curr. Biol. 2007, 17, 1055–1060. [Google Scholar] [CrossRef] [Green Version]
- Searle, I.; He, Y.; Turck, F.; Vincent, C.; Fornara, F.; Kröber, S.; Amasino, R.A.; Coupland, G. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 2006, 20, 898–912. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.R.G.; Ai, X.Y.; Zhang, J.Z. Genetic regulation of flowering time in annual and perennial plants. Wiley Interdiscip. Rev. RNA 2014, 5, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Kemi, U.; Niittyvuopio, A.; Toivainen, T.; Pasanen, A.; Quilot-Turion, B.; Holm, K.; Lagercrantz, U.; Savolainen, O.; Kuittinen, H. Role of vernalization and of duplicated FLOWERING LOCUS C in the perennial Arabidopsis lyrata. N. Phytol. 2013, 197, 323–335. [Google Scholar] [CrossRef]
- Wang, R.; Farrona, S.; Vincent, C.; Joecker, A.; Schoof, H.; Turck, F.; Alonso-Blanco, C.; Coupland, G.; Albani, M.C. PEP1 regulates perennial flowering in Arabis alpina. Nature 2009, 459, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Z.; Li, Z.M.; Mei, L.; Yao, J.L.; Hu, C.G. PtFLC homolog from trifoliate orange (Poncirus trifoliata) is regulated by alternative splicing and experiences seasonal fluctuation in expression level. Planta 2009, 229, 847–859. [Google Scholar] [CrossRef]
- An, H.; Jiang, S.; Zhang, J.; Xu, F.; Zhang, X. Comparative transcriptomic analysis of differentially expressed transcripts associated with flowering time of loquat (Eriobotya japonica lindl.). Horticulturae 2021, 7, 171. [Google Scholar] [CrossRef]
- Zhou, C.M.; Zhang, T.Q.; Wang, X.; Yu, S.; Lian, H.; Tang, H.; Feng, Z.Y.; Zozomova-Lihová, J.; Wang, J.W. Molecular basis of age-dependent vernalization in Cardamine flexuosa. Science 2013, 340, 1097–1100. [Google Scholar] [CrossRef]
- Sheng, F.; Hu, B.; Jin, Q.; Wang, J.; Wu, C.; Luo, Z. The analysis of phenolic compounds in walnut husk and pellicle by UPLC-Q-Orbitrap HRMS and HPLC. Molecules 2021, 26, 3013. [Google Scholar] [CrossRef]
- Bernard, A.; Lheureux, F.; Dirlewanger, E. Walnut: Past and future of genetic improvement. Tree Genet. Genomes 2018, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Quan, S.; Niu, J.; Zhou, L.; Xu, H.; Ma, L.; Qin, Y. Stages identifying and transcriptome profiling of the floral transition in Juglans regia. Sci. Rep. 2019, 9, 7092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassankhah, A.; Rahemi, M.; Ramshini, H.; Sarikhani, S.; Vahdati, K. Flowering in Persian walnut: Patterns of gene expression during flower development. BMC Plant Biol. 2020, 20, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrano, A.; Britton, M.; Zaini, P.A.; Zimin, A.V.; Workman, R.E.; Puiu, D.; Bianco, L.; Di Pierro, E.A.; Allen, B.J.; Chakraborty, S.; et al. High-quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. Gigascience 2020, 9, giaa050. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Friedländer, M.R.; MacKowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Xiaochen, B.; Wang, S. TargetFinder: A software for antisense oligonucleotide target site selection based on MAST and secondary structures of target mRNA. Bioinformatics 2005, 21, 1401–1402. [Google Scholar]
- Wigge, P.A. FT, A mobile developmental signal in plants. Curr. Biol. 2011, 21, R374–R378. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, N. LEAFY, a pioneer transcription factor in plants: A mini-review. Front. Plant Sci. 2021, 12, 1274. [Google Scholar] [CrossRef]
- Kimura, Y.; Aoki, S.; Ando, E.; Kitatsuji, A.; Watanabe, A.; Ohnishi, M.; Takahashi, K.; Inoue, S.I.; Nakamichi, N.; Tamada, Y.; et al. A flowering integrator, SOC1, affects stomatal opening in Arabidopsis thaliana. Plant Cell Physiol. 2015, 56, 640–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahdati, K.; Mohseniazar, M. Early bearing genotypes of walnut: A suitable material for breeding and high density orchards. Acta Horti. 2016, 1139, 101–105. [Google Scholar] [CrossRef]
- Breton, C.; Cornu, D.; Chriqui, D.; Sauvanet, A.; Capelli, P.; Germain, E.; Jay-Allemand, C. Somatic embryogenesis, micropropagation and plant regeneration of “Early Mature” walnut trees (Juglans regia) that flower in vitro. Tree Physiol. 2004, 24, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.Y.; Zhang, Q.X.; Song, X.B.; Zha, W.W.; Pei, D. Analysis on key factors affecting somatic embryogenesis and plant regeneration in precocious walnuts. For. Res. 2019, 32, 34–39. [Google Scholar]
- Vahdati, K.; Razaee, R.; Mirmasoomi, M. Micropropagation of some dwarf and early mature walnut genotypes. Biotechnology 2009, 8, 171–175. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, P.; Liu, Q.; Wei, Z.; Riaz, A.; Chachar, S.; Gu, X. Rice homolog of Sin3-associated polypeptide 30, OsSFL1, mediates histone deacetylation to regulate flowering time during short days. Plant Biotechnol. J. 2020, 18, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fu, X.; Wang, Y.; Liu, R.; He, Y. Polycomb-mediated gene silencing by the BAH–EMF1 complex in plants. Nat. Genet. 2018, 50, 1254–1261. [Google Scholar] [CrossRef]
- Liao, X.; Li, Y.; Hu, Z.; Lin, Y.; Zheng, B.; Ding, J. Poplar acetylome profiling reveals lysine acetylation dynamics in seasonal bud dormancy release. Plant. Cell Environ. 2021, 44, 1830–1845. [Google Scholar] [CrossRef]
- Agustí, M.; Mesejo, C.; Muñoz-Fambuena, N.; Vera-Sirera, F.; de Lucas, M.; Martínez-Fuentes, A.; Reig, C.; Iglesias, D.J.; Primo-Millo, E.; Blázquez, M.A. Fruit-dependent epigenetic regulation of flowering in Citrus. New Phytol. 2020, 225, 367–384. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Q.; Mo, R.; Chen, W.; Zhang, Q.; Sheng, F.; Wu, C.; Zhang, R.; Luo, Z. Identification and Comparative Analysis of Genes and MicroRNAs Involved in the Floral Transition of the Xinjiang Early-Flowering Walnut (Juglans regia L.). Horticulturae 2022, 8, 136. https://doi.org/10.3390/horticulturae8020136
Jin Q, Mo R, Chen W, Zhang Q, Sheng F, Wu C, Zhang R, Luo Z. Identification and Comparative Analysis of Genes and MicroRNAs Involved in the Floral Transition of the Xinjiang Early-Flowering Walnut (Juglans regia L.). Horticulturae. 2022; 8(2):136. https://doi.org/10.3390/horticulturae8020136
Chicago/Turabian StyleJin, Qiang, Rongli Mo, Wenxing Chen, Qinglin Zhang, Fang Sheng, Cuiyun Wu, Rui Zhang, and Zhengrong Luo. 2022. "Identification and Comparative Analysis of Genes and MicroRNAs Involved in the Floral Transition of the Xinjiang Early-Flowering Walnut (Juglans regia L.)" Horticulturae 8, no. 2: 136. https://doi.org/10.3390/horticulturae8020136