
Global Methylation Analysis Using MSAP Reveals Differences in Chilling-Associated DNA Methylation Changes during Dormancy Release in Contrasting Sweet Cherry Varieties
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, Chilling Requirement (CR), and Growing Degree Hours (GDH) Estimation
2.2. Genomic DNA Extraction
2.3. Methylation-Sensitive Amplified Polymorphism (MSAP) Assay
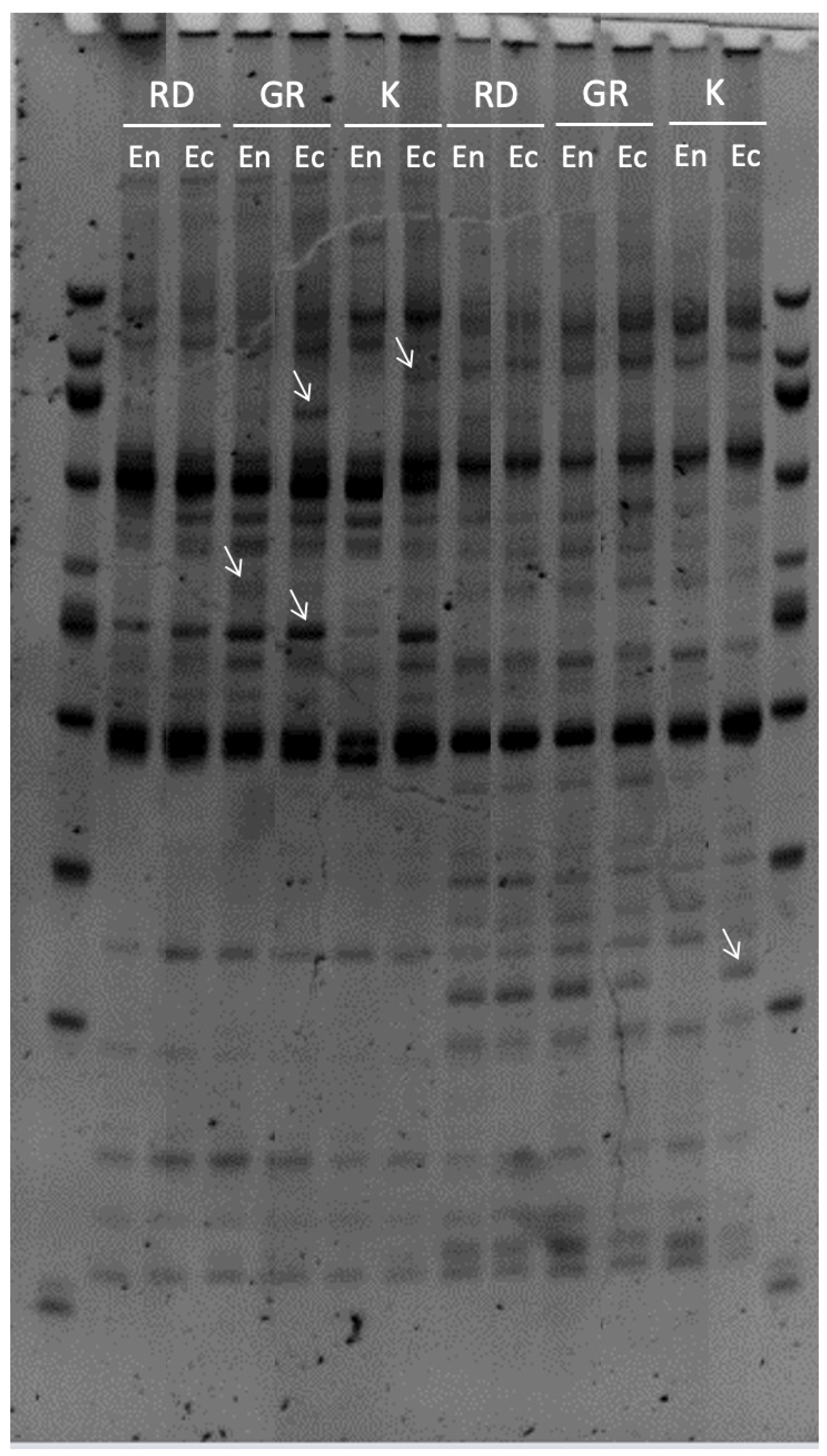
2.4. MSAP Bands Selection
2.5. Library Construction
2.6. Sequencing and Bioinformatics Analysis of MSAP Bands
2.7. MethylC-Seq
3. Results
3.1. Chilling Requirement Determination
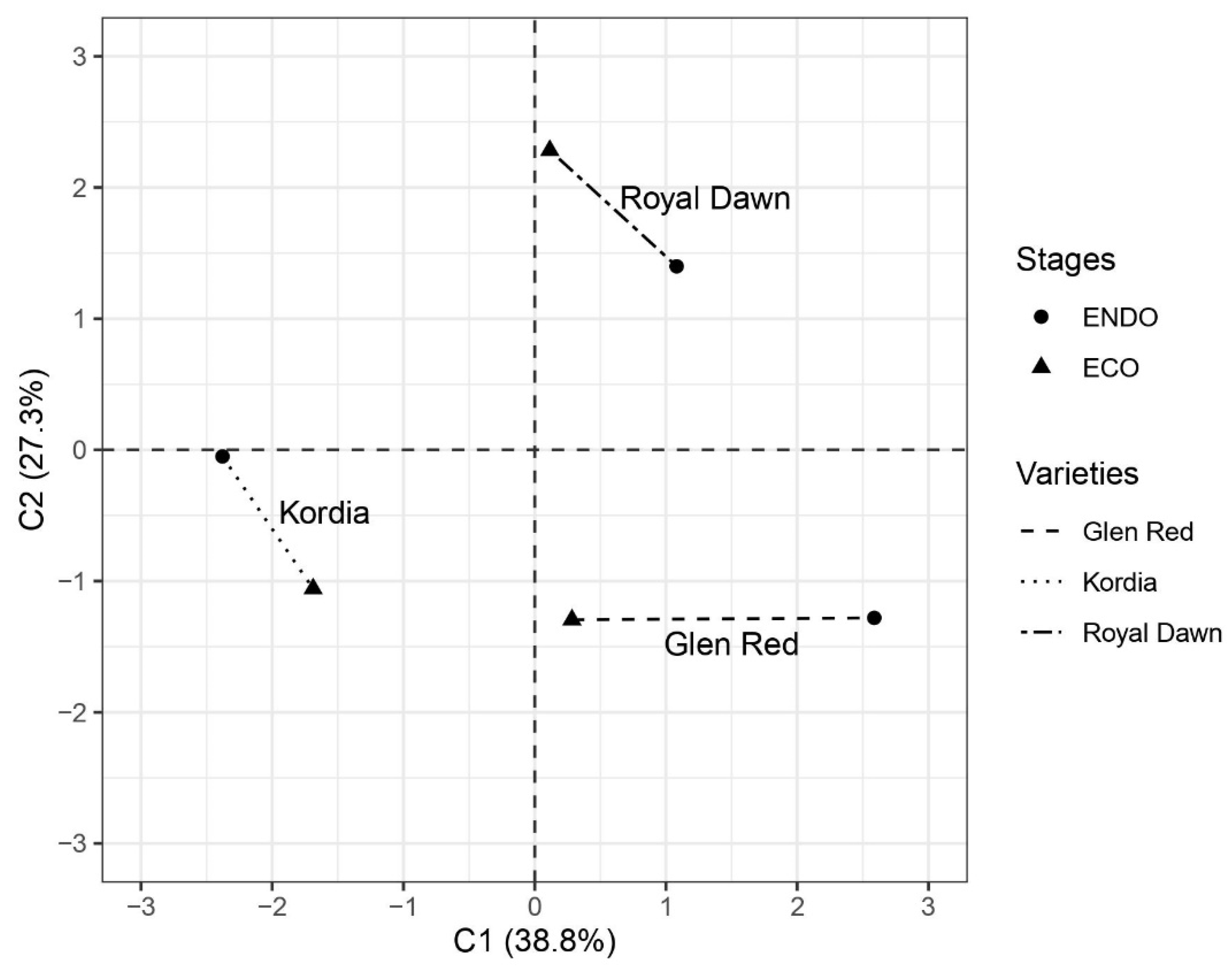
3.2. Methylation Profiling of Sweet Cherry Floral Buds at Different Dormancy Stages
3.3. Dynamics of Methylation/Demethylation Events Concerning Dormancy Stage Transition
3.4. Sequence Analysis of MSAP Polymorphic Fragments
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Schoot, C.; Rinne, P.L. Dormancy cycling at the shoot apical meristem, Transitioning between self-organization and self-arrest. Plant Sci. 2011, 180, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.A. Dormancy, a new universal terminology. HortScience 1987, 22, 817–820. [Google Scholar] [CrossRef]
- Albuquerque, N.; García-Montiel, F.; Carrillo, A.; Burgos, L. Chilling and heat requirements of sweet cherry cultivars and the relationship between altitude and the probability of satisfying the chill requirements. Environ. Exp. Bot. 2008, 64, 162–170. [Google Scholar] [CrossRef]
- Castede, S.; Campoy, J.A.; Quero-García, J.; Dantec, L.L.; Lafarge, M.; Barreneche, T.; Wenden, B.; Dirlewanger, E. Genetic determinism of phenological traits highly affected by climate change in Prunus avium, flowering date dissected into chilling and heat requirements. New Phytol. 2014, 202, 703–715. [Google Scholar] [CrossRef]
- Campoy, J.; Darbyshire, R.; Dirlewanger, E.; Quero-Garcia, J.; Wenden, B. Yield potential definition of the chilling requirement reveals likely underestimation of the risk of climate change on winter chill accumulation. Int. J. Biometeorol. 2019, 63, 183–192. [Google Scholar] [CrossRef]
- Santamaría, M.E.; Hasbún, R.; Valera, M.J.; Meijón, M.; Valledor, L.; Rodríguez, J.L.; Toorop, P.E.; Cañal, M.J.; Rodríguez, R. Acetylated H4 histone and genomic DNA methylation patterns during bud set and bud burst in Castanea sativa. Plant Physiol. 2009, 166, 1360–1369. [Google Scholar] [CrossRef]
- Ríos, G.; Leída, C.; Conejero, A.; Badenes, M.L. Epigenetic regulation of bud dormancy events in perennial Plants. Front. Plant Sci. 2014, 5, 247. [Google Scholar]
- Kumar, G.; Kumari, U.; Kumar, A. Chilling-mediated DNA methylation changes during dormancy and its release reveal the importance of epigenetic regulation during winter dormancy in Apple (Malus x domestica Borkh.). PLoS ONE 2016, 11, e0149934. [Google Scholar]
- Rothkegel, K.; Sánchez, E.; Montes, C.; Greve, M.; Tapia, S.; Bravo, S.; Prieto, H.; Almeida, A.M. DNA methylation and small interference RNAs participate in the regulation of MADS-box genes involved in dormancy in sweet cherry (Prunus avium L.). Tree Physiol. 2017, 12, 1739–1751. [Google Scholar] [CrossRef] [Green Version]
- Rothkegel, K.; Sandoval, P.; Ulloa, L.; Riveros, A.; Lillo-Carmona, V.; Cáceres-Molina, J.; Almeida, A.M.; Meneses, C. Dormant but active: Chilling accumulation modulates the epigenome and transcriptome of Prunus avium during bud dormancy. Front. Plant Sci. 2021, 11, 1115. [Google Scholar] [CrossRef]
- Prudencio, A.S.; Werner, O.; Martínez-García, P.J.; Dicenta, F.; Ros, R.M.; Martínez-Gómez, P. DNA methylation analysis of dormancy release in almond (Prunus dulcis) flower buds using epi-genotyping by sequencing. Int. J. Mol. Sci. 2018, 19, 3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing; maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 2014–2220. [Google Scholar] [CrossRef] [PubMed]
- Bonduriansky, R.; Crean, A.J.; Day, T. The implications of nongenetic inheritance for evolution in changing environments. Evol. Appl. 2012, 5, 192–201. [Google Scholar] [CrossRef]
- Avramidou, E.; Ganopoulos, I.; Doulis, A.; Tsaftaris, A.; Aravanopoulos, F. Beyond population genetics, natural epigenetic variation in wild cherry (Prunus avium). Tree Genet. Genomes 2015, 11, 95. [Google Scholar] [CrossRef]
- Guevara, M.Á.; de María, N.; Sáez-Laguna, E.; Vélez, M.D.; Cervera, M.T.; Cabezas, J.A. Analysis of DNA cytosine methylation patterns using methylation-sensitive amplification polymorphism (MSAP). In Plant Epigenetics. Methods in Molecular Biology; Kovalchuck, I., Ed.; Humana Press: Boston, MA, USA, 2017; Volume 1456. [Google Scholar]
- Cao, D.H.; Gao, X.; Liu, J.; Kimatu, J.N.; Geng, S.J.; Wang, X.P.; Zhao, J.; Shi, D.C. Methylation sensitive amplified polymorphism (MSAP) reveals that alkali stress triggers more DNA hypomethylation levels in cotton (Gossypium hirsutum L.) roots than salt stress. Afr. J. Biotechnol. 2011, 10, 18971–18980. [Google Scholar]
- Shan, X.; Wang, X.; Yang, G.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Yuan, Y. Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J. Plant Biol. 2013, 56, 32–38. [Google Scholar] [CrossRef]
- Tang, X.M.; Tao, X.; Wang, Y.; Ma, D.W.; Li, D.; Yang, H.; Ma, X.R. Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol. Genet. Genom. 2014, 289, 1075–1084. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, X.; Wang, H.; Shi, F.; Liu, Y.; Liu, J.; Li, L.; Wang, D.; Liu, B. Cytosine methylation alteration in natural populations of Leymus chinensis induced by multiple abiotic stresses. PLoS ONE 2013, 8, e55772. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, P.; Orlowska, R.; Niedziela, A. A relative quantitative methylation-sensitive amplified polymorphism (MSAP) method for the analysis of abiotic stress. BMC Plant Biol. 2017, 17, 79. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, J.H. Chilling requirements of peach varieties. J. Am. Soc. Hortic. Sci. 1950, 56, 122–128. [Google Scholar]
- Richardson, E.; Seeley, S.; Walker, D. A model for estimating the completion of rest for ‘Redhaven’ and ‘Elberta’ peach trees. HortScience 1974, 9, 331–332. [Google Scholar] [CrossRef]
- Hasbún, R.; Iturra, C.; Moraga, P.; Wachtendorff, P.; Quiroga, P.; Valenzuela, S. An efficient and reproducible protocol for production of AFLP markers in tree genomes using fluorescent capillary detection. Tree Genet. Genomes 2012, 8, 925–931. [Google Scholar] [CrossRef]
- Rodríguez-López, C.; Morán, P.; Lago, F.C.; Espiñeira, M.; Beckmann, M.; Consuegra, S. Detection and quantification of tissue of origin in salmon and veal products using Methylation Sensitive AFLPs. Food Chem. 2012, 131, 1493–1498. [Google Scholar] [CrossRef]
- Karan, R.; DeLeon, T.; Biradar, H.; Subudhi, P. Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 2012, 7, e40203. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Quesada, H.; Rolán-Alvarez, E. Impact of amplified fragment length polymorphism size homoplasy on the estimation of population genetic diversity and the detection of selective loci. Genetics 2008, 179, 539–554. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Figueroa, A. Msap, a tool for the statistical analysis of methylation-sensitive amplified polymorphim data. Mol. Ecol. Resour. 2013, 13, 522–527. [Google Scholar] [CrossRef]
- Shirasawa, K.; Isuzugawa, K.; Ikenaga, M.; Saito, Y.; Yamamoto, T.; Hirakawa, H.; Isobe, S. The genome sequence of sweet cherry (Prunus avium) for use in genomics-assisted breeding. DNA Res. 2017, 24, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E.; Paris, R.; Aramini, V.; Gazza, L.; Rossini, L.; et al. The Peach v2.0 release: High-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 2017, 18, 225. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [Green Version]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Kimatu, J.N.; Xu, K.; Liu, B. DNA cytosine methylation in plant development. J. Genet. Genom. 2010, 37, 1–12. [Google Scholar] [CrossRef]
- Zluvova, J.; Janousek, B.; Vyskot, B. Immunohistochemical study of DNA methylation dynamics during plant development. J. Exp. Bot. 2001, 52, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Steimer, A.; Schob, H.; Grossniklaus, U. Epigenetic control of plant development, new layers of complexity. Curr. Opin. Plant Biol. 2004, 7, 11–19. [Google Scholar] [CrossRef]
- Valledor, L.; Hasbún, R.; Meijón, M.; Rodríguez, J.L.; Santamaría, E.; Viejo, M.; Berdasco, M.; Feito, I.; Fraga, M.; Cañal, M.J.; et al. Involvement of DNA methylation in tree development and micropropagation. Plant Cell Tissue Organ Cult. 2007, 91, 75–86. [Google Scholar] [CrossRef]
- Kuhn, N.; Maldonado, J.; Ponce, C.; Arellano, M.; Time, A.; Multari, S.; Martens, S.; Carrera, E.; Donoso, J.M.; Sagredo, B.; et al. RNAseq reveals different transcriptomic responses to GA3 in early and midseason varieties before ripening initiation in sweet cherry fruits. Sci. Rep. 2021, 11, 13075. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.L.; Tucker, M.R.; Khor, S.F.; Shirley, N.; Lahnstein, J.; Beahan, C.; Bacic, A.; Burton, R.A. Differences in glycosyltransferase family 61 accompany variation in seed coat mucilage composition in Plantago spp. J. Exp. Bot. 2016, 67, 6481–6495. [Google Scholar] [CrossRef] [Green Version]
- Cosgrove, D.J. Plant cell wall extensibility, connecting plant cell growth with cell wall structure; mechanics; and the action of wall-modifying enzymes. J. Exp. Bot. 2016, 67, 463–476. [Google Scholar] [CrossRef] [Green Version]
- Kofler, J.; Milyaev, A.; Würtz, B.; Pfannstiel, J.; Flachowsky, H.; Wünsche, J.-N. Proteomic differences in apple spur buds from high and non-cropping trees during floral initiation. J. Proteom. 2022, 253, 104459. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, L.; Chen, M.; Fang, X.; Xie, Z.; Gong, P.; Huang, Y.; Wang, Z.; Fang, J. Comparative transcriptome analysis provides insight into regulation pathways and temporal and spatial expression characteristics of grapevine (Vitis vinifera) dormant buds in different nodes. BMC Plant Biol. 2020, 20, 390. [Google Scholar] [CrossRef]
- Shani, Z.; Dekel, M.; Roiz, L.; Horowitz, M.; Kolosovski, N.; Lapidot, S.; Alkan, S.; Koltai, H.; Tsabary, G.; Goren, R.; et al. Expression of endo-1;4-beta-glucanase (cel1) in Arabidopsis thaliana is associated with plant growth; xylem development and cell wall thickening. Plant Cell Rep 2006, 25, 1067–1074. [Google Scholar] [CrossRef]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Friters, A.; Pot, J.; Paleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef] [Green Version]
- Kaity, A.; Ashmore, S.E.; Drew, R.A.; Duloo, M.E. Assessment of genetic and epigenetic changes following cryopreservation in papaya. Plant Cell Rep. 2008, 27, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- López, C.M.R.; Wetten, A.C.; Wilkinson, M.J. Progressive erosion of genetic and epigenetic variation in callus-derived cocoa (Theobroma cacao) plants. New Phytol. 2010, 186, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Landey, R.B.; Cenci, A.; Georget, F.; Bertrand, B.; Camayo, G.; Dechamp, E.; Herrera, J.C.; Santoni, S.; Lashermes, P.; Simpson, J.; et al. High genetic and epigenetic stability in Coffea arabica plants derived from embryogenic suspensions and secondary embryogenesis as revealed by AFLP, MSAP and the phenotypic variation rate. PLoS ONE 2013, 8, e56372. [Google Scholar]



{kind=link}
{kind=link}
| Stages | Budbreak (%) | ‘Royal Dawn’ | ‘Glen Red’ | ‘Kordia’ |
|---|---|---|---|---|
| ENDO | 0% | 173 CH | 173 CH | 1127 CH |
| ECO | ≥50% | 516 CH | 684 CH | 1637 CH |
| ‘Royal Dawn’ | ‘Glen Red’ | ‘Kordia’ | ||||
|---|---|---|---|---|---|---|
| Band Types (Quantity) | ENDO | ECO | ENDO | ECO | ENDO | ECO |
| Type I | 65 | 65 | 55 | 67 | 72 | 76 |
| Type II | 23 | 24 | 27 | 19 | 20 | 20 |
| Type III | 13 | 13 | 20 | 13 | 14 | 11 |
| Type IV | 20 | 19 | 19 | 22 | 15 | 14 |
| Total methylation (%) | 46.3 | 46.3 | 54.5 | 44.6 | 40.5 | 37.1 |
| Fully methylated bands (%) | 27.2 | 26.4 | 32.2 | 28.9 | 23.9 | 20.6 |
| Hemi-methylated bands (%) | 19.0 | 19.8 | 22.3 | 15.7 | 16.5 | 16.5 |
| Banding Pattern | Bands Detected | ||||||
|---|---|---|---|---|---|---|---|
| ENDO | ECO | ‘Royal Dawn’ | ‘Glen Red’ | ‘Kordia’ | |||
| HpaII | MspI | HpaII | MspI | ENDO/ECO | ENDO/ECO | ENDO/ECO | |
| No changes | |||||||
| A | 1 | 1 | 1 | 1 | 60 | 55 | 71 |
| B | 1 | 0 | 1 | 0 | 11 | 10 | 10 |
| C | 0 | 1 | 0 | 1 | 19 | 18 | 13 |
| D | 0 | 0 | 0 | 0 | 18 | 16 | 13 |
| 108 (89.2 *) | 99 (95.8) | 111 (91.7) | |||||
| Demethylation | |||||||
| E | 1 | 0 | 1 | 1 | 1 | 3 | 4 |
| F | 0 | 1 | 1 | 1 | 4 | 6 | 0 |
| G | 0 | 0 | 1 | 1 | 0 | 0 | 1 |
| H | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| 7 (5.8) | 15 (12.4) | 7 (5.8) | |||||
| Methylation | |||||||
| K | 1 | 1 | 1 | 0 | 1 | 0 | 1 |
| L | 1 | 1 | 0 | 1 | 3 | 0 | 1 |
| M | 1 | 0 | 0 | 1 | 1 | 1 | 0 |
| N | 1 | 1 | 0 | 0 | 1 | 0 | 0 |
| O | 1 | 0 | 0 | 0 | 0 | 6 | 0 |
| P | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| 6 (5) | 7 (5.8) | 3 (2.5) | |||||
| Shannon Index | ||||||
|---|---|---|---|---|---|---|
| Stages | # MSL | # NML | % Polym MSL | % Polym NML | MSL | NML |
| ENDO | 61 | 60 | 41 | 22 | 0.641 | 0.637 |
| ECO | 48 | 73 | 44 | 22 | 0.650 | 0.637 |
| All stages | 66 | 55 | 35 | 11 | 0.596 | 0.620 |
| Bands | Selective Primers Combination | Methylation Status Change Detected by MSAP | Variety | Gene ID | Probable Function | E-Value | Differential Methylated Regions (DMR) Fold Change (FC) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Methylation Pattern * | ENDO | ECO | Royal Dawn 516 CH vs. 0 | Kordia 1637 CH vs. 0 | ||||||
| M1 | ATA/TCCA | N | non-methylated | methylated | Kordia | Pav_sc0000350.1_g1030.1.mk | Glycosyltransferase 61. Gibberelin-regulated protein 9. uncharacterized protein LOC18787986 [Prunus persica] > gi|1139755066|gb|ONH90523.1| hypothetical protein PRUPE_8G059200 [Prunus persica] | 0 | na † | na |
| M2 | ATA/TCCA | C | methylated | methylated | Royal Dawn | Pav_sc0002774.1_g020.1.br | PREDICTED: uncharacterized protein LOC103331076 | 1.75 × 10−43 | −2.18 | na |
| Pav_sc0000008.1_g540.1.br | hypothetical protein PRUPE_ppa020796mg | 1.75 × 10-43 | −1.72 | na | ||||||
| Pav_sc0000287.1_g500.1.br | uncharacterized protein LOC103336567 | 2.91 × 10-28 | −2.66 | na | ||||||
| M3 | ACT/TCCA | G | methylated | non-methylated | Kordia | Pav_sc0000507.1_g110.1.mk | No description | 3.50 × 10 −11 | 1.87 | 1.01 |
| M4 | ACG/TAG | F | methylated | non-methylated | Kordia | Pav_sc0001502.1_g550.1.br | cyclic nucleotide-gated ion channel 1-like | 3.62094 × 10−08 | −1.97 | na |
| M5 | ACG/TAG | A | non-methylated | non-methylated | Kordia | Pav_sc0000131.1_g230.1.mk | Endo-1,4-beta-glucanase | 0 | 1.56 | na |
| M6 | ACG/TAG | J | methylated | hemimethylated | Glen Red | Pav_sc0005790.1_g010.1.br | PREDICTED: probable serine/threonine-protein kinase abkC | 1.66 × 10 −59 | 1.10 | −2.99 |
| Pav_sc0005156.1_g020.1.br | PREDICTED: tether containing UBX domain for GLUT4 | 1.66 × 10 −59 | −2.95 | na | ||||||
| Pav_sc0000323.1_g460.1.mk | Pyruvate kinase, cytosolic isozyme OS = Nicotiana tabacum PE = 2 SV = 1 | 8.94 × 10−36 | na | −3.06 | ||||||
| M7 | ACG/TAG | C | methylated | methylated | Kordia | Pav_sc0005790.1_g010.1.br | PREDICTED: probable serine/threonine-protein kinase abkC | 1.84 × 10−44 | 1.10 | −2.99 |
| Pav_sc0005156.1_g010.1.mk | PREDICTED: tether containing UBX domain for GLUT4 | 1.84 × 10 −44 | −2.95 | na | ||||||
| Pav_sc0000323.1_g460.1.mk | Pyruvate kinase, cytosolic isozyme OS = Nicotiana tabacum PE = 2 SV = 1 | 6.93 × 10 −33 | na | −3.06 | ||||||
| M8 | ATA/TCG | B | methylated | methylated | Kordia | Pav_sc0000933.1_g020.1.br | predicted protein | 4.54 × 10 −150 | 3.69 | −1.13 |
| Pav_sc0001012.1_g020.1.br | PREDICTED: uncharacterized protein LOC103328417 | 4.54 × 10−150 | −1.39 | −1.11 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narváez, G.; Muñoz-Espinoza, C.; Soto, E.; Rothkegel, K.; Bastías, M.; Gutiérrez, J.; Bravo, S.; Hasbún, R.; Meneses, C.; Almeida, A.M. Global Methylation Analysis Using MSAP Reveals Differences in Chilling-Associated DNA Methylation Changes during Dormancy Release in Contrasting Sweet Cherry Varieties. Horticulturae 2022, 8, 962. https://doi.org/10.3390/horticulturae8100962
Narváez G, Muñoz-Espinoza C, Soto E, Rothkegel K, Bastías M, Gutiérrez J, Bravo S, Hasbún R, Meneses C, Almeida AM. Global Methylation Analysis Using MSAP Reveals Differences in Chilling-Associated DNA Methylation Changes during Dormancy Release in Contrasting Sweet Cherry Varieties. Horticulturae. 2022; 8(10):962. https://doi.org/10.3390/horticulturae8100962
Chicago/Turabian StyleNarváez, Gabriela, Claudia Muñoz-Espinoza, Esteban Soto, Karin Rothkegel, Macarena Bastías, José Gutiérrez, Soraya Bravo, Rodrigo Hasbún, Claudio Meneses, and Andrea Miyasaka Almeida. 2022. "Global Methylation Analysis Using MSAP Reveals Differences in Chilling-Associated DNA Methylation Changes during Dormancy Release in Contrasting Sweet Cherry Varieties" Horticulturae 8, no. 10: 962. https://doi.org/10.3390/horticulturae8100962