
Tailoring the Properties of Ni(111)/Graphone Interfaces by Intercalation of Al and Na: A DFT Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
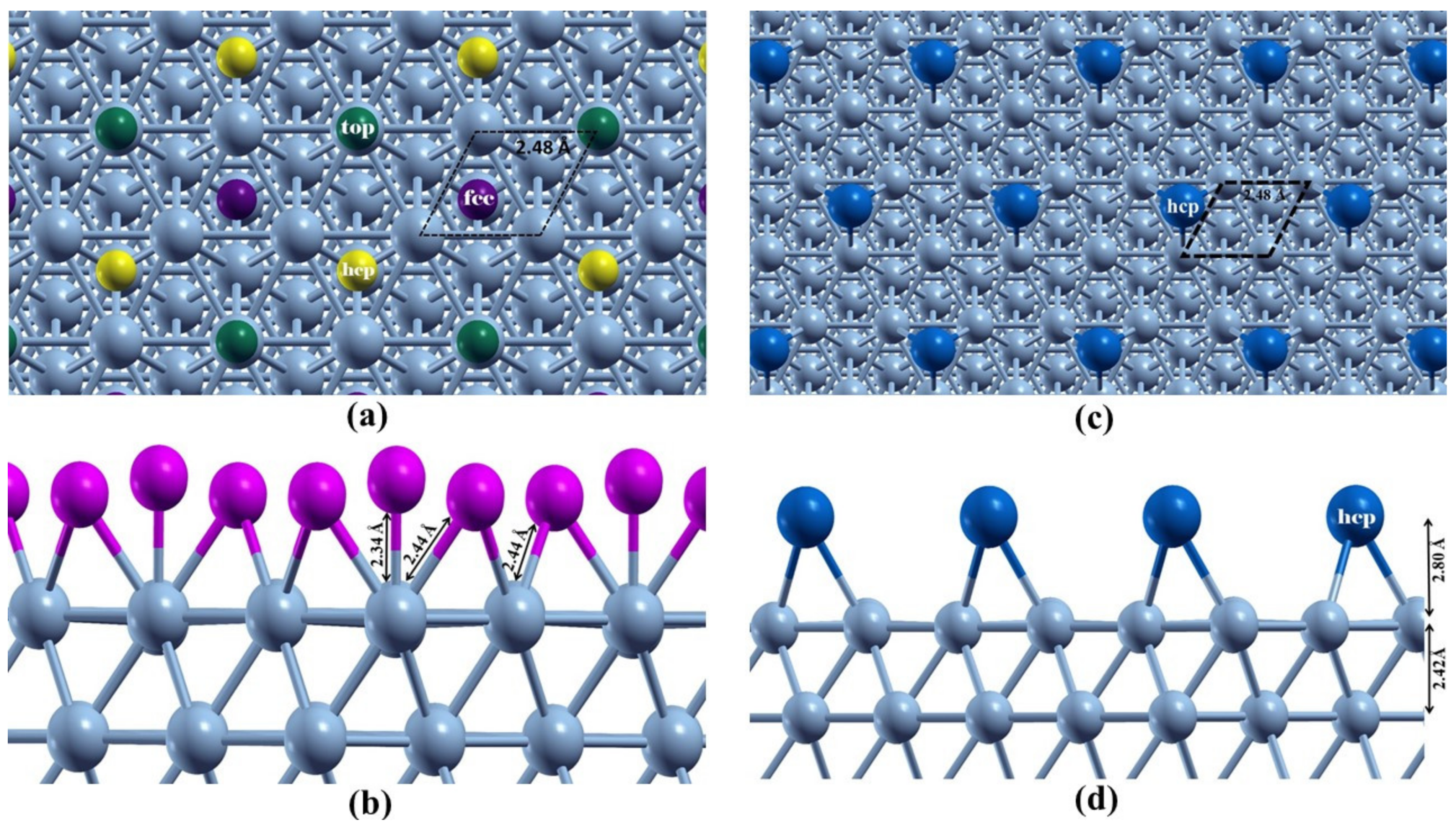
3.1. Modified Ni(111) Surface
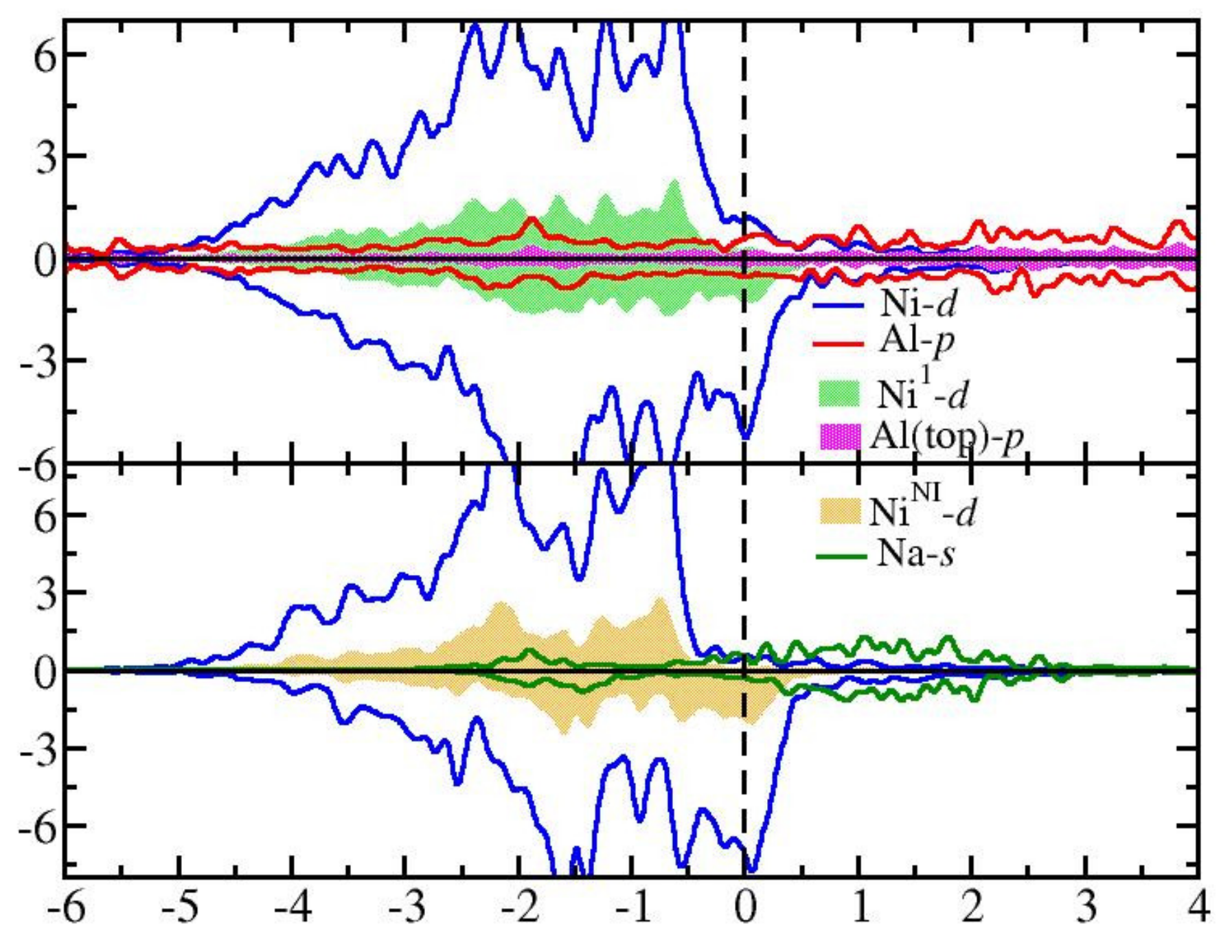
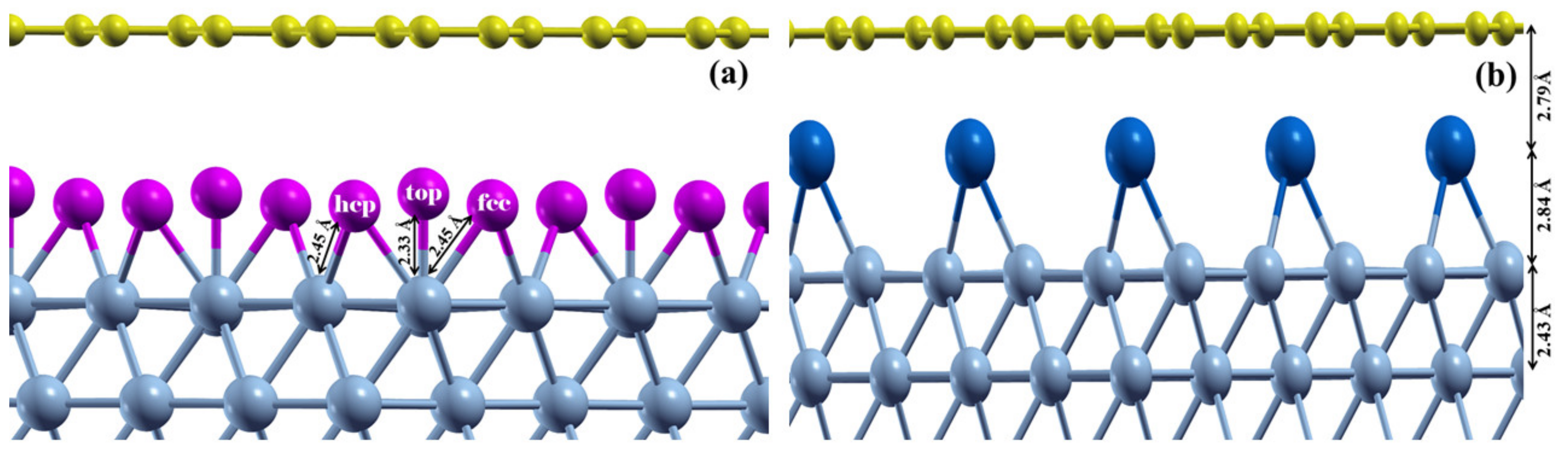
3.2. Graphene Adsorbed on Ni(111)/Al and Ni(111)/Na Surfaces
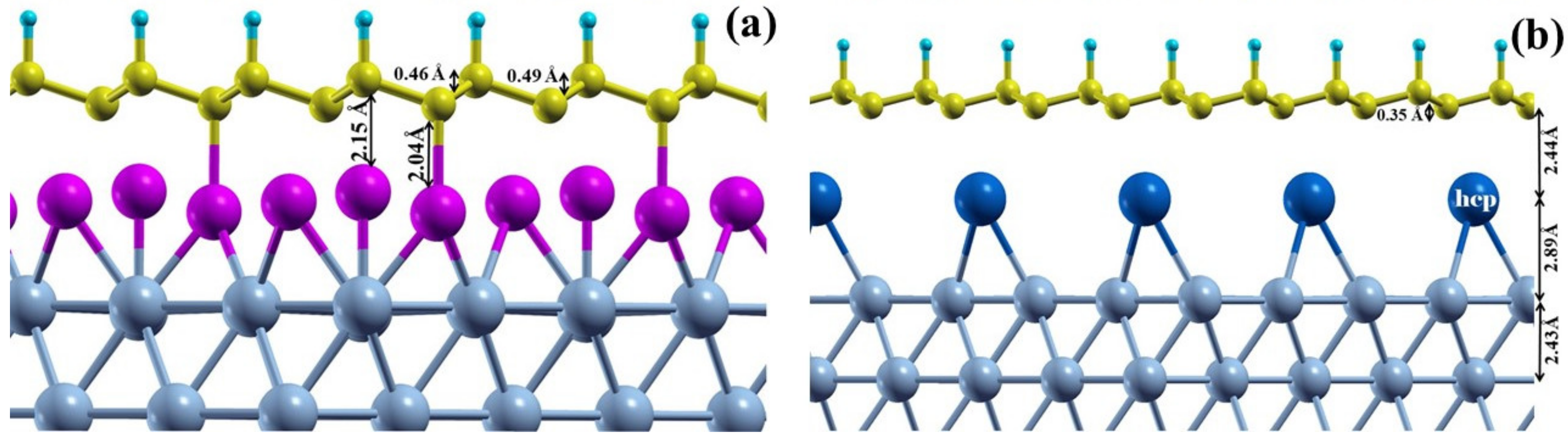
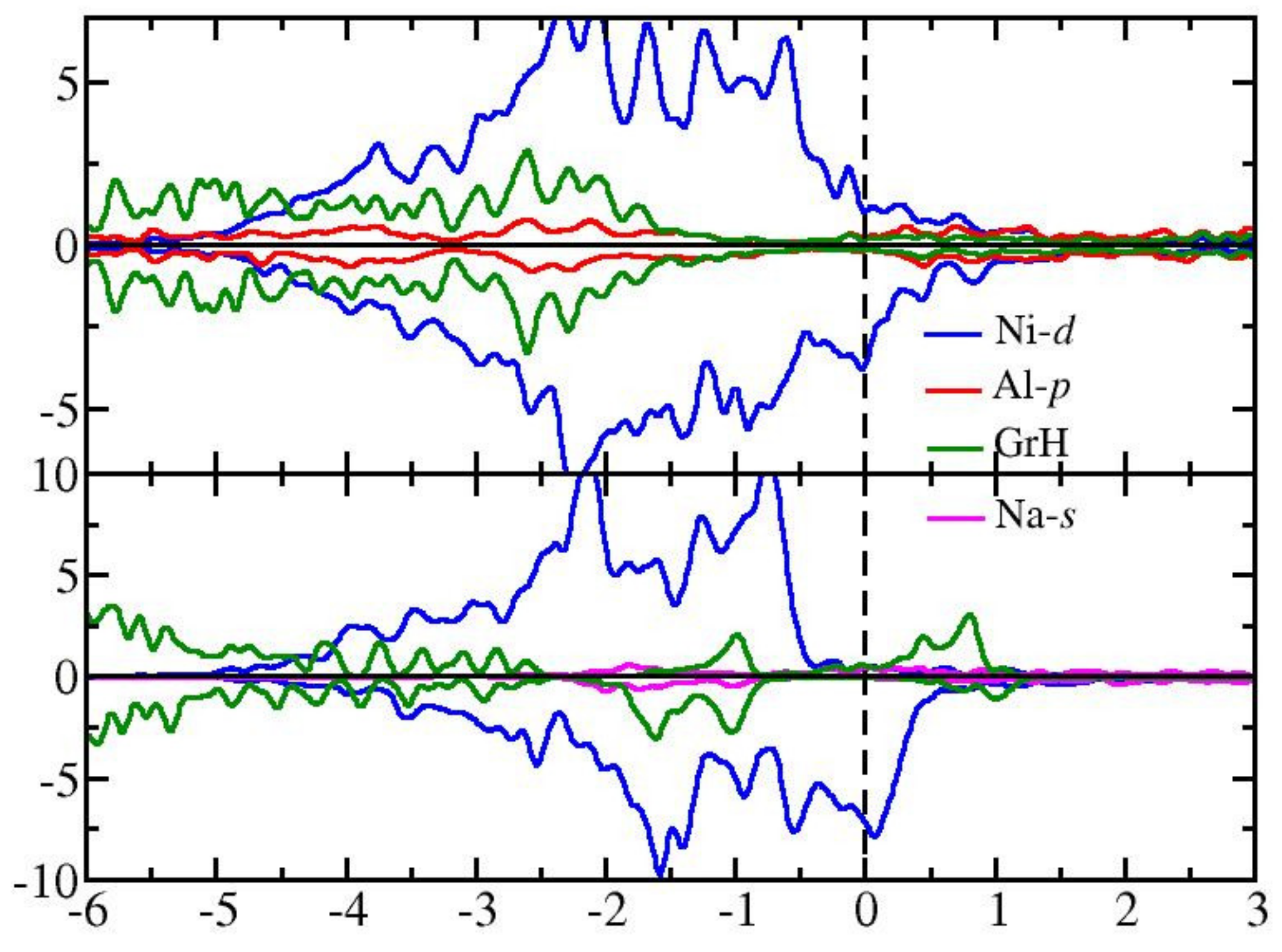
3.3. Graphone on the Modified Ni(111) Surface
4. Discussions
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Englert, J.M.; Dotzer, C.; Yang, G.; Schmid, M.; Papp, C.; Gottfried, J.M.; Steinrück, H.P.; Spiecker, E.; Hauke, F.; Hirsch, A. Covalent bulk functionalization of graphene. Nat. Chem. 2011, 3, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Brzhezinskaya, M.; Kononenko, O.; Matveev, V.; Zotov, A.; Khodos, I.I.; Levashov, V.; Volkov, V.; Bozhko, S.I.; Chekmazov, S.V.; Roshchupkin, D. Engineering of Numerous Moiré Superlattices in Twisted Multilayer Graphene for Twistronics and Straintronics Applications. ACS Nano 2021, 15, 12358–12366. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Otyepka, M.; Bourlinos, A.B.; Chandra, V.; Kim, N.; Kemp, K.C.; Hobza, P.; Zboril, R.; Kim, K.S. Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications. Chem. Rev. 2012, 112, 6156–6214. [Google Scholar] [CrossRef]
- Sofo, J.O.; Chaudhari, A.S.; Barber, G.D. Graphane: A two-dimensional hydrocarbon. Phys. Rev. B Condens. Matter. Mater. Phys. 2007, 75, 153401. [Google Scholar] [CrossRef] [Green Version]
- Shnitov, V.V.; Rabchinskii, M.K.; Brzhezinskaya, M.; Stolyarova, D.Y.; Pavlov, S.V.; Baidakova, M.V.; Shvidchenko, A.V.; Kislenko, V.A.; Kislenko, S.A.; Brunkov, P.N. Valence Band Structure Engineering in Graphene Derivatives. Small 2021, 17, 2104316. [Google Scholar] [CrossRef]
- ZZhang, W.; Lu, W.-C.; Zhang, H.-X.; Ho, K.; Wang, C. Hydrogen adatom interaction on graphene: A first-principles study. Carbon 2018, 131, 137–141. [Google Scholar] [CrossRef]
- Brzhezinskaya, M.; Shmatko, V.; Yalovega, G.; Krestinin, A.; Bashkin, I.; Bogoslavskaja, E. Electronic Structure of Hydrogenated Carbon Nanotubes Studied by Core Level Spectroscopy. J. Electron. Spectrosc. Relat. Phenom. 2014, 196, 99. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Q.; Sun, Q.; Chen, X.S.; Kawazoe, Y.; Jena, P. Ferromagnetism in semihydrogenated graphene sheet. Nano Lett. 2009, 9, 3867–3870. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, W.X. The structure and magnetism of graphone. AIP Adv. 2012, 2, 042138. [Google Scholar] [CrossRef]
- Šljivančanin, Ž.; Balog, R.; Hornekær, L. Magnetism in graphene induced by hydrogen adsorbates. Chem. Phys. Lett. 2012, 541, 70–74. [Google Scholar] [CrossRef]
- Brzhezinskaya, M.; Belenkov, E.A.; Greshnyakov, V.A.; Yalovega, G.E.; Bashkin, I.O. New aspects in the study of carbon-hydrogen interaction in hydrogenated carbon nanotubes for energy storage applications. J. Alloys Compd. 2019, 792, 713–720. [Google Scholar] [CrossRef]
- Morse, J.R.; Zugell, D.A.; Patterson, E.; Baldwin, J.W.; Willauer, H.D. Hydrogenated graphene: Important material properties regarding its application for hydrogen storage. J. of Power Sources 2021, 494, 229734. [Google Scholar] [CrossRef]
- Zhao, W.; Gebhardt, J.; Späth, F.; Gotterbarm, K.; Gleichweit, C.; Steinrück, H.P.; Görling, A.; Papp, C. Reversible hydrogenation of graphene on Ni(111)-synthesis of graphone. Chem.—Eur. J. 2015, 21, 3347–3358. [Google Scholar] [CrossRef]
- Sutter, P.; Sadowski, J.T.; Sutter, E.A. Chemistry under cover: Tuning metal-graphene interaction by reactive intercalation. J. Am. Chem. Soc. 2010, 132, 8175–8179. [Google Scholar] [CrossRef]
- Pervan, P.; Lazić, P. Adsorbed or intercalated: Na on graphene/Ir(111). Phys. Rev. Mater. 2017, 1, 044202. [Google Scholar] [CrossRef] [Green Version]
- Karpan, V.M.; Khomyakov, P.A.; Starikov, A.A.; Giovannetti, G.; Zwierzycki, M.; Talanana, M.; Brocks, G.; Van Den Brink, J.; Kelly, P.J. Theoretical prediction of perfect spin filtering at interfaces between close-packed surfaces of Ni or Co and graphite or graphene. Phys. Rev. B 2008, 78, 195419. [Google Scholar] [CrossRef] [Green Version]
- Halle, J.; Néel, N.; Kröger, J. Filling the Gap: Li-Intercalated Graphene on Ir(111). J. Phys. Chem. C 2016, 120, 5067–5073. [Google Scholar] [CrossRef]
- Weser, M.; Voloshina, E.N.; Horn, K.; Dedkov, Y.S. Electronic structure and magnetic properties of the graphene/Fe/Ni(111) intercalation-like system. Phys. Chem. Chem. Phys. 2011, 13, 7534–7539. [Google Scholar] [CrossRef] [Green Version]
- Praveen, C.S.; Piccinin, S.; Fabris, S. Adsorption of alkali adatoms on graphene supported by the Au/Ni(111) surface. Phys. Rev. B Condens. Matter Mater. Phys. 2015, 92, 075403. [Google Scholar] [CrossRef]
- Joshi, N.; Gaurav, C.; Ballav, N.; Ghosh, P. Tuning electronic and magnetic properties of the graphone/Ni(111) interface by oxygen intercalation: A first-principles prediction. Phys. Rev. B 2020, 101, 195401. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Vanderbilt, D. Communications Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; de Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.S.; Park, J.H.; Hwang, H.N.; Laishram, T.S.; Kim, K.S.; Kang, M.H.; Hwang, C.C. Quasi-free-standing graphene monolayer on a Ni crystal through spontaneous Na intercalation. Phys. Rev. X 2014, 4, 031016. [Google Scholar] [CrossRef] [Green Version]
- Voloshina, E.N.; Generalov, A.; Weser, M.; Böttcher, S.; Horn, K.; Dedkov, Y.S. Structural and electronic properties of the graphene/Al/Ni(111) intercalation system. New. J. Phys. 2011, 13, 113028. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Archana, R.; Joshi, N.; Raja, T. Tailoring the Properties of Ni(111)/Graphone Interfaces by Intercalation of Al and Na: A DFT Study. C 2022, 8, 62. https://doi.org/10.3390/c8040062
Archana R, Joshi N, Raja T. Tailoring the Properties of Ni(111)/Graphone Interfaces by Intercalation of Al and Na: A DFT Study. C. 2022; 8(4):62. https://doi.org/10.3390/c8040062
Chicago/Turabian StyleArchana, Ramakrishnan, Niharika Joshi, and Thirumalaiswamy Raja. 2022. "Tailoring the Properties of Ni(111)/Graphone Interfaces by Intercalation of Al and Na: A DFT Study" C 8, no. 4: 62. https://doi.org/10.3390/c8040062