

Insights into Online microRNA Bioinformatics Tools


Abstract
:1. Introduction
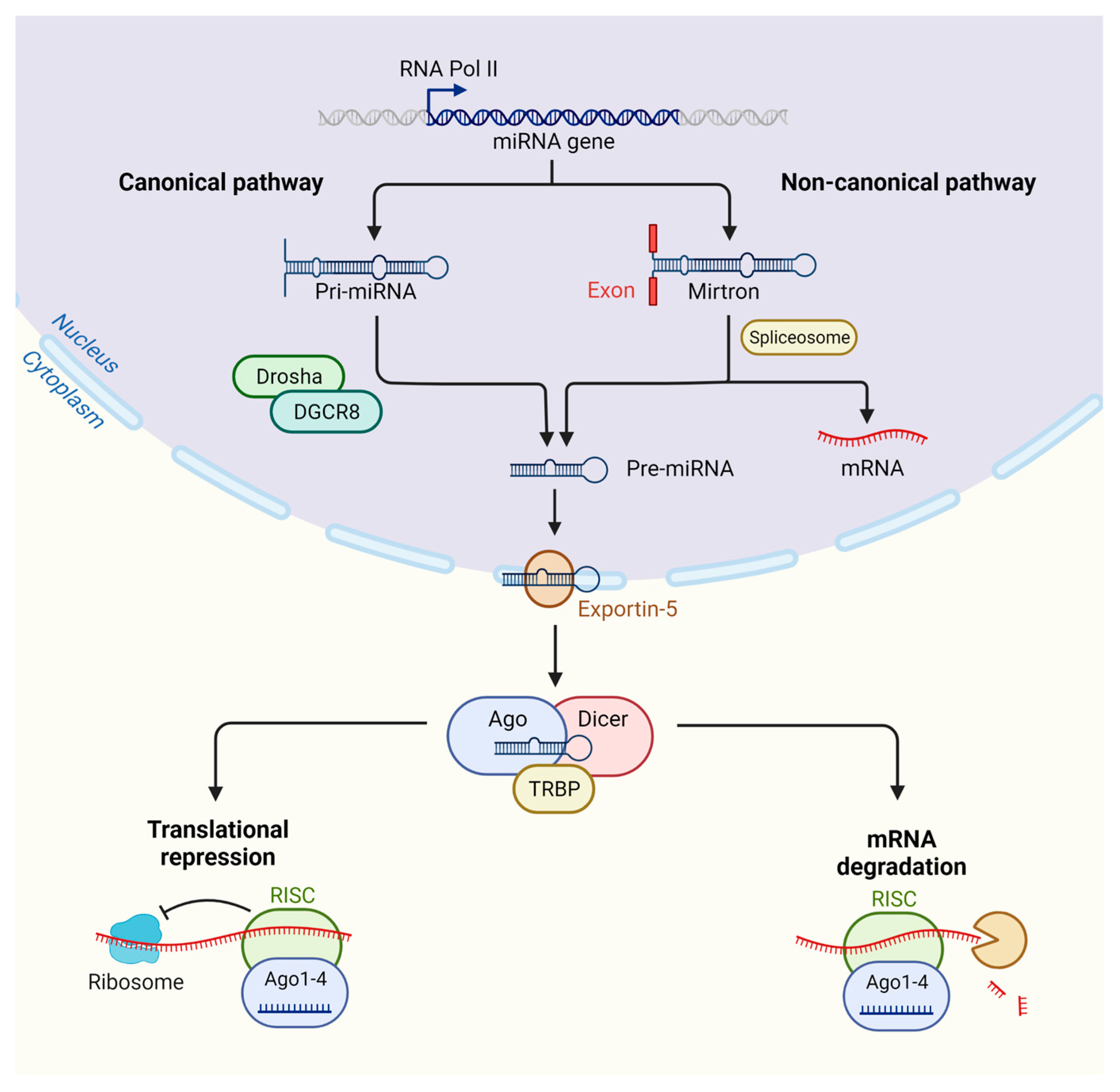
2. MicroRNA Biogenesis and Targeting
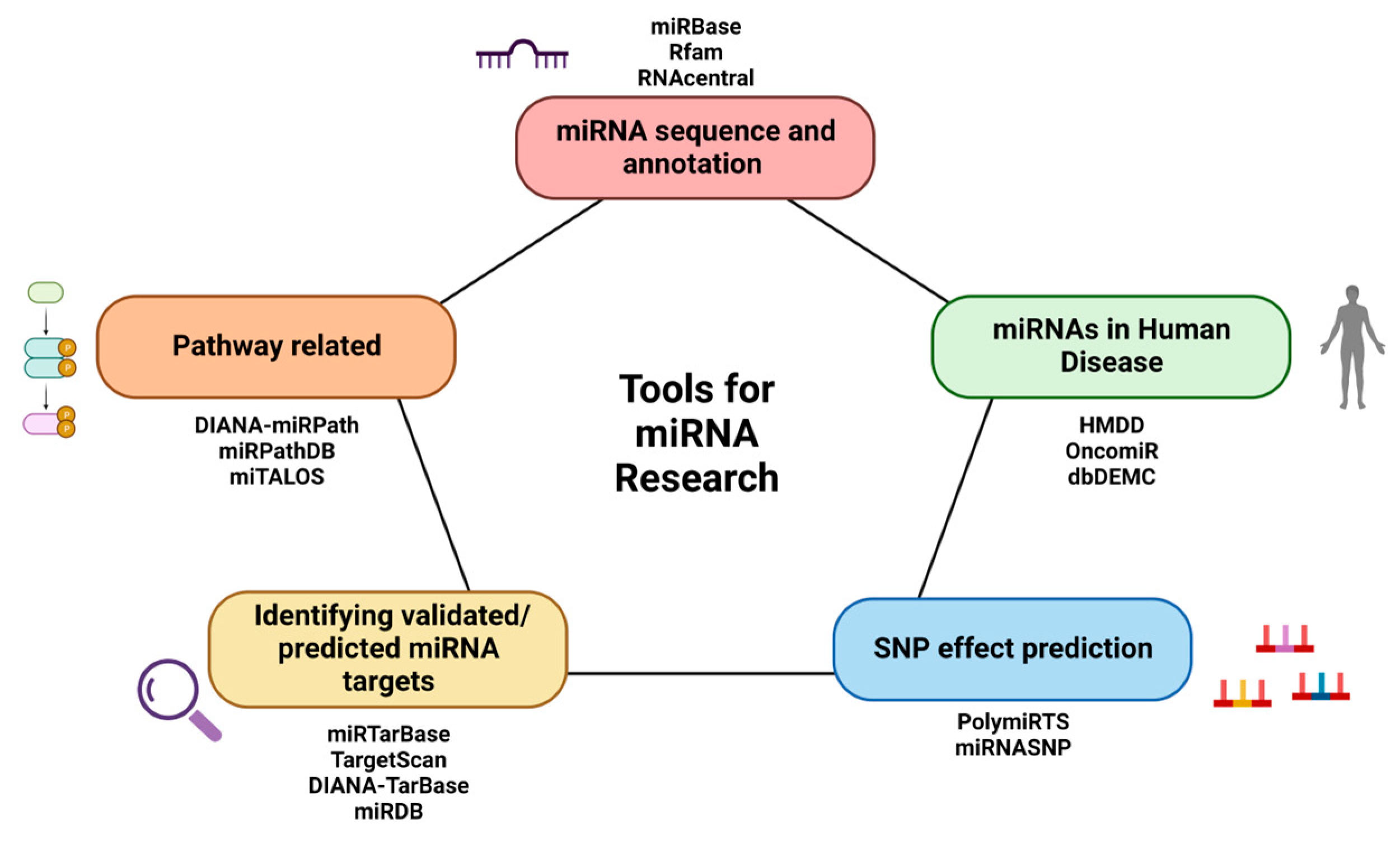
3. Tools for miRNA Sequences and Annotations
4. Tools for the Identification of Validated/Predicted miRNA Targets
5. Tools Related to Human Diseases
6. Tools for Pathway Identification
7. Tools for SNP Effect Prediction
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [Green Version]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grässer, F.A.; van Dyk, L.F.; Kiong Ho, C.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Stavast, C.J.; Erkeland, S.J. The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells 2019, 8, 1465. [Google Scholar] [CrossRef] [Green Version]
- Berezikov, E.; Chung, W.J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian Mirtron Genes. Mol. Cell 2007, 28, 328–336. [Google Scholar] [CrossRef] [Green Version]
- de Rooij, L.A.; Mastebroek, D.J.; ten Voorde, N.; van der Wall, E.; van Diest, P.J.; Moelans, C.B. The microRNA Lifecycle in Health and Cancer. Cancers 2022, 14, 5748. [Google Scholar] [CrossRef]
- Titov, I.I.; Vorozheykin, P.S. Comparing miRNA structure of mirtrons and non-mirtrons. BMC Genom. 2018, 19, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, M.T.; Czaplinski, K.; Görlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear Export of MicroRNA Precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, S.W.; Bass, B.L. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 2001, 293, 2269–2271. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, R.; Han, B.W.; Hung, J.H.; Xu, J.; Weng, Z.; Zamore, P.D. Dicer partner proteins tune the length of mature miRNAs in flies and mammals. Cell 2012, 151, 533–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meijer, H.A.; Smith, E.M.; Bushell, M. Regulation of miRNA strand selection: Follow the leader? Biochem. Soc. Trans. 2014, 42, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Wahle, E.; Winkler, G.S. RNA decay machines: Deadenylation by the Ccr4-Not and Pan2-Pan3 complexes. Biochim. Biophys. Acta 2013, 1829, 561–570. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.H.; Ghoshal, K.; Villén, J.; Bartel, D.P. MRNA Destabilization Is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rocca, G.; Olejniczak, S.H.; González, A.J.; Briskin, D.; Vidigal, J.A.; Spraggon, L.; DeMatteo, R.G.; Radler, M.R.; Lindsten, T.; Ventura, A.; et al. In vivo, Argonaute-bound microRNAs exist predominantly in a reservoir of low molecular weight complexes not associated with mRNA. Proc. Natl. Acad. Sci. USA 2015, 112, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 2018, 25, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Santovito, D.; Weber, C. Non-canonical features of microRNAs: Paradigms emerging from cardiovascular disease. Nat. Rev. Cardiol. 2022, 19, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y. Long Non-Coding RNAs. Methods and Protocols. Anticancer. Res. 2016, 36, 2044–2045. [Google Scholar]
- Keller, A.; Gröger, L.; Tschernig, T.; Solomon, J.; Laham, O.; Schaum, N.; Wagner, V.; Kern, F.; Schmartz, G.P.; Li, Y.; et al. MiRNATissueAtlas2: An update to the human miRNA tissue atlas. Nucleic Acids Res. 2022, 50, D211–D221. [Google Scholar] [CrossRef]
- Huang, Y.; Zou, Q.; Wang, S.P.; Tang, S.M.; Zhang, G.Z.; Shen, X.J. The discovery approaches and detection methods of microRNAs. Mol. Biol. Rep. 2011, 38, 4125–4135. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Zhuo, Y.; Gao, G.; Shi, J.A.; Zhou, X.; Wang, X. MiRNAs: Biogenesis, origin and evolution, functions on virus-host interaction. Cell. Physiol. Biochem. 2013, 32, 499–510. [Google Scholar] [CrossRef]
- Riolo, G.; Cantara, S.; Marzocchi, C.; Ricci, C. miRNA targets: From prediction tools to experimental validation. Methods Protoc. 2021, 4, 1. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Saini, H.K.; Van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, 154–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. MiRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.D.; Bateman, A.; Petrov, A.I. Non-Coding RNA Analysis Using the Rfam Database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Sweeney, B.A.; Petrov, A.I.; Ribas, C.E.; Finn, R.D.; Bateman, A.; Szymanski, M.; Karlowski, W.M.; Seemann, S.E.; Gorodkin, J.; Cannone, J.J.; et al. RNAcentral 2021: Secondary structure integration, improved sequence search and new member databases. Nucleic Acids Res. 2021, 49, D212–D220. [Google Scholar] [CrossRef]
- The RNAcentral Consortium RNAcentral: A hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019, 47, D221–D229. [CrossRef] [PubMed] [Green Version]
- Da Hsu, S.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. MiRTarBase: A database curates experimentally validated microRNA-target interactions. Nucleic Acids Res. 2011, 39, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Da Hsu, S.; Tseng, Y.T.; Shrestha, S.; Lin, Y.L.; Khaleel, A.; Chou, C.H.; Chu, C.F.; Huang, H.Y.; Lin, C.M.; Ho, S.Y.; et al. MiRTarBase update 2014: An information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014, 42, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.H.; Chang, N.W.; Shrestha, S.; Da Hsu, S.; Lin, Y.L.; Lee, W.H.; Yang, C.D.; Hong, H.C.; Wei, T.Y.; Tu, S.J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. MiRTarBase update 2018: A resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.D.; Li, J.; Huang, K.Y.; Shrestha, S.; Hong, H.C.; Tang, Y.; Chen, Y.G.; Jin, C.N.; Yu, Y.; et al. MiRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.Y.; Lin, Y.C.D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. MiRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of Mammalian MicroRNA Targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Grimson, A.; Farh, K.K.H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Vergoulis, T.; Vlachos, I.S.; Alexiou, P.; Georgakilas, G.; Maragkakis, M.; Reczko, M.; Gerangelos, S.; Koziris, N.; Theodore, D.; Hatzigeorgiou, A.G. TarBase 6.0: Capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2012, 40, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Chen, G.; Cui, Q. Towards the understanding of microRNA and environmental factor interactions and their relationships to human diseases. Sci. Rep. 2012, 2, 318. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.W.; Chen, Y.; Chen, S.; Wang, X. OncomiR: An online resource for exploring pan-cancer microRNA dysregulation. Bioinformatics 2018, 34, 713–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y.; et al. DbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Wang, Y.; Ling, Y.; Zhou, C.; Wang, H.; Teschendorff, A.E.; Zhao, Y.; Zhao, H.; He, Y.; Zhang, G.; et al. dbDEMC 3.0: Functional Exploration of Differentially Expressed miRNAs in Cancers of Human and Model Organisms. Genom. Proteom. Bioinform. 2022, 20, 446–454. [Google Scholar] [CrossRef]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef]
- Backes, C.; Kehl, T.; Stöckel, D.; Fehlmann, T.; Schneider, L.; Meese, E.; Lenhof, H.P.; Keller, A. MiRPathDB: A new dictionary on microRNAs and target pathways. Nucleic Acids Res. 2017, 45, D90–D96. [Google Scholar] [CrossRef]
- Kehl, T.; Kern, F.; Backes, C.; Fehlmann, T.; Stöckel, D.; Meese, E.; Lenhof, H.P.; Keller, A. MiRPathDB 2.0: A novel release of the miRNA Pathway Dictionary Database. Nucleic Acids Res. 2020, 48, D142–D147. [Google Scholar] [CrossRef]
- Kowarsch, A.; Preusse, M.; Marr, C.; Theis, F.J. miTALOS: Analyzing the tissue-specific regulation of signaling pathways by human and mouse microRNAs. RNA 2012, 18, 1101. [Google Scholar] [CrossRef] [Green Version]
- Preusse, M.; Theis, F.J.; Mueller, N.S. miTALOS v2: Analyzing tissue specific microRNA function. PLoS ONE 2016, 11, e0151771. [Google Scholar] [CrossRef]
- Bao, L.; Zhou, M.; Wu, L.; Lu, L.; Goldowitz, D.; Williams, R.W.; Cui, Y. PolymiRTS Database: Linking polymorphisms in microRNA target sites with complex traits. Nucleic Acids Res. 2007, 35, 51–54. [Google Scholar] [CrossRef] [Green Version]
- Ziebarth, J.D.; Bhattacharya, A.; Chen, A.; Cui, Y. PolymiRTS database 2.0: Linking polymorphisms in microRNA target sites with human diseases and complex traits. Nucleic Acids Res. 2012, 40, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, 86–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.; Tong, Y.; Zhang, H.-M.; Guo, A.-Y. miRNASNP: A database of miRNA related SNPs and their effects on miRNA function. BMC Bioinform. 2012, 13, 2105. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Liu, C.; Liu, W.; Wu, Y.; Ma, Z.; Chen, H.; Guo, A.Y. An update of miRNASNP database for better SNP selection by GWAS data, miRNA expression and online tools. Database 2015, 2015, bav029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.J.; Fu, X.; Xia, M.; Zhang, Q.; Gu, Z.; Guo, A.Y. MiRNASNP-v3: A comprehensive database for SNPs and disease-related variations in miRNAs and miRNA targets. Nucleic Acids Res. 2021, 49, D1276–D1281. [Google Scholar] [CrossRef]
- Riffo-Campos, Á.L.; Riquelme, I.; Brebi-Mieville, P. Tools for sequence-based miRNA target prediction: What to choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.C.; Bovolenta, L.A.; Nachtigall, P.G.; Herkenhoff, M.E.; Lemke, N.; Pinhal, D. Combining results from distinct microRNA target prediction tools enhances the performance of analyses. Front. Genet. 2017, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, Q.; Zhang, J.; Li, C.; Miao, Y.R.; Lei, Q.; Li, Q.; Guo, A.Y. EVmiRNA: A database of miRNA profiling in extracellular vesicles. Nucleic Acids Res. 2019, 47, D89–D93. [Google Scholar] [CrossRef] [Green Version]
- Marceca, G.P.; Distefano, R.; Tomasello, L.; Lagana, A.; Russo, F.; Calore, F.; Romano, G.; Bagnoli, M.; Gasparini, P.; Ferro, A.; et al. MiREDiBase, a manually curated database of validated and putative editing events in microRNAs. Sci. Data 2021, 8, 199. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buniello, A.; Macarthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.P.; George, N.; et al. Expression Atlas: Gene and protein expression across multiple studies and organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kowdley, K.V. MicroRNAs in Common Human Diseases. Genom. Proteom. Bioinform. 2012, 10, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Ren, N.; Wang, M. microRNA-212-induced protection of the heart against myocardial infarction occurs via the interplay between AQP9 and PI3K/Akt signaling pathway. Exp. Cell Res. 2018, 370, 531–541. [Google Scholar] [CrossRef]
- Liu, J.Y.; Shang, J.; Mu, X.D.; Gao, Z.Y. Protective effect of down-regulated microRNA-27a mediating high thoracic epidural block on myocardial ischemia-reperfusion injury in mice through regulating ABCA1 and NF-κB signaling pathway. Biomed. Pharmacother. 2019, 112, 108606. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargutkar, N.; Hariharan, P.; Nadkarni, A. Dynamic interplay of microRNA in diseases and therapeutic. Clin. Genet. 2023, 103, 268–276. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Rodney Brister, J.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef]
- Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Hartline, E.; et al. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [CrossRef] [Green Version]
- Huntley, R.P.; Kramarz, B.; Sawford, T.; Umrao, Z.; Kalea, A.; Acquaah, V.; Martin, M.J.; Mayr, M.; Lovering, R.C. Expanding the horizons of microRNA bioinformatics. RNA 2018, 24, 1005–1017. [Google Scholar] [CrossRef]
- Garcia-Moreno, A.; Carmona-Saez, P. Computational methods and software tools for functional analysis of mirna data. Biomolecules 2020, 10, 1252. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochim. Biophys. Acta—Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Kramarz, B.; Lovering, R.C. Gene Ontology: A Resource for Analysis and Interpretation of Alzheimer’s Disease Data. In Alzheimer’s Disease; Codon Publications: Singapore, 2019; pp. 23–36. ISBN 9780646809687. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Fehlmann, T.; Kern, F.; Kehl, T.; Lenhof, H.P.; Meese, E.; Keller, A. MiRCarta: A central repository for collecting miRNA candidates. Nucleic Acids Res. 2018, 46, D160–D167. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Yuan, Y.; Weidhaas, J.B. Functional microRNA binding site variants. Mol. Oncol. 2019, 13, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Saunders, M.A.; Liang, H.; Li, W.H. Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. USA 2007, 104, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Cammaerts, S.; Strazisar, M.; De Rijk, P.; Del Favero, J. Genetic variants in microRNA genes: Impact on microRNA expression, function, and disease. Front. Genet. 2015, 6, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machowska, M.; Galka-Marciniak, P.; Kozlowski, P. Consequences of genetic variants in miRNA genes. Comput. Struct. Biotechnol. J. 2022, 20, 6443–6457. [Google Scholar] [CrossRef] [PubMed]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Roberts, J.T.; Borchert, G.M. Computational prediction of microRNA target genes, target prediction databases, and web resources. Methods Mol. Biol. 2017, 1617, 109–122. [Google Scholar] [CrossRef]
- Akhtar, M.M.; Micolucci, L.; Islam, M.S.; Olivieri, F.; Procopio, A.D. Bioinformatic tools for microRNA dissection. Nucleic Acids Res. 2016, 44, 24–44. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Category | Tool Name | Organism | Last Update | Summary Features | URL | References |
|---|---|---|---|---|---|---|
| miRNA sequences and annotation | miRBase | 271 species, including humans | 2019 |
| https://www.mirbase.org/ | [32,33,34,35] |
| Rfam | 9 species, including humans | 2022 |
| https://rfam.xfam.org/ | [36,37] | |
| RNAcentral | >800,00 species, including humans | 2022 |
| https://rnacentral.org/ | [38,39] | |
| Target Discovery | miRTarBase | 37 species, including humans | 2021 |
| https://mirtarbase.cuhk.edu.cn/ | [40,41,42,43,44,45] |
| TargetScan | 13 species, including humans | 2018 |
| https://www.targetscan.org/vert_72/ | [46,47,48,49] | |
| DIANA-TarBase | 18 species, including humans | 2017 |
| https://dianalab.e-ce.uth.gr/html/diana/web/index.php?r=tarbasev8 | [50,51] | |
| miRDB | 5 species, including humans | 2019 |
| http://mirdb.org/ | [52,53] | |
| Human disease-related | HMDD | Humans | 2019 |
| http://www.cuilab.cn/hmdd | [54,55,56,57] |
| OncomiR | Humans | 2017 |
| http://www.oncomir.org/ | [58] | |
| dbDEMC | 3 species, including humans | 2021 |
| https://www.biosino.org/dbDEMC/index | [59,60] | |
| Pathway-related | DIANA-miRPath | 7 species, including humans | 2015 |
| https://dianalab.e-ce.uth.gr/html/mirpathv3/index.php?r=mirpath | [61] |
| miRPathDB | 2 species, including humans | 2018 |
| https://mpd.bioinf.uni-sb.de/ | [62,63] | |
| miTALOS | 2 species, including humans | 2016 |
| http://mips.helmholtz-muenchen.de/mitalos/#/search | [64,65] | |
| SNP effect prediction | PolymiRTS | 2 species, including humans | 2014 |
| https://compbio.uthsc.edu/miRSNP// | [66,67,68] |
| miRNASNP | Humans | 2020 |
| http://bioinfo.life.hust.edu.cn/miRNASNP/#!/ | [69,70,71] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luna Buitrago, D.; Lovering, R.C.; Caporali, A. Insights into Online microRNA Bioinformatics Tools. Non-Coding RNA 2023, 9, 18. https://doi.org/10.3390/ncrna9020018
Luna Buitrago D, Lovering RC, Caporali A. Insights into Online microRNA Bioinformatics Tools. Non-Coding RNA. 2023; 9(2):18. https://doi.org/10.3390/ncrna9020018
Chicago/Turabian StyleLuna Buitrago, Diana, Ruth C. Lovering, and Andrea Caporali. 2023. "Insights into Online microRNA Bioinformatics Tools" Non-Coding RNA 9, no. 2: 18. https://doi.org/10.3390/ncrna9020018