


Drying of Hierarchically Organized Porous Silica Monoliths–Comparison of Evaporative and Supercritical Drying
 ,
,
Abstract
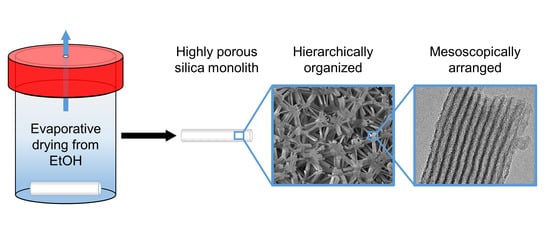
:
1. Introduction
2. Results and Discussion
2.1. Linear Shrinkage and Densities
2.2. Structural Characteristics
3. Conclusions
4. Materials and Methods
4.1. Chemicals
4.2. Synthesis of Hierarchically Ordered Silica Gels
4.3. Drying
4.4. Characterization
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hayase, G.; Kugimiya, K.; Ogawa, M.; Kodera, Y.; Kanamori, K.; Nakanishi, K.J. The thermal conductivity of polymethylsilsesquioxane aerogels and xerogels with varied pore sizes for practical application as thermal superinsulators. Mater. Chem. A 2014, 2, 6525–6531. [Google Scholar] [CrossRef]
- Nakanishi, K.; Tanaka, N. Sol–Gel with Phase Separation. Hierarchically Porous Materials Optimized for High-Performance Liquid Chromatography Separations. Acc. Chem. Res. 2007, 40, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Sachse, A.; Galarneau, A.; Fajula, F.; Di Renzo, F.; Creux, P.; Coq, B. Functional silica monoliths with hierarchical uniform porosity as continuous flow catalytic reactors. Microporous Mesoporous Mater. 2011, 140, 58–68. [Google Scholar] [CrossRef]
- Tan, I.; Zarafshani, Z.; Lutz, J.-F.; Titirici, M.-M. PEGylated Chromatography: Efficient Bioseparation on Silica Monoliths Grafted with Smart Biocompatible Polymers. ACS Appl. Mater. Interfaces 2009, 1, 1869–1872. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, A.; Sachse, A.; Said, B.; Pélisson, C.-H.; Boscaro, P.; Brun, N.; Courtheoux, L.; Olivi-Tran, N.; Coasne, B.; Fajula, F. Hierarchical porous silica monoliths: A novel class of microreactors for process intensification in catalysis and adsorption. C. R. Chimie 2016, 19, 231–247. [Google Scholar] [CrossRef]
- Maleki, H.; Huesing, N. Current status, opportunities and challenges in catalytic and photocatalytic applications of aerogels: Environmental protection aspects. Appl. Catal. B 2018, 221, 530–555. [Google Scholar] [CrossRef]
- Ko, Y.G.; Lee, H.J.; Kim, J.Y.; Choi, U.S. Hierarchically Porous Aminosilica Monolith as a CO2 Adsorbent. ACS Appl. Mater. Interfaces 2014, 6, 12988–12996. [Google Scholar] [CrossRef]
- Wilson, S.M.W.; Gabriel, V.A.; Tezel, F.H. Adsorption of components from air on silica aerogels. Microporous Mesoporous Mater. 2020, 305, 110297. [Google Scholar] [CrossRef]
- Enke, D.; Glaeser, R.; Tallarek, U. Sol–Gel and Porous Glass-Based Silica Monoliths with Hierarchical Pore Structure for Solid-Liquid Catalysis. Chem. Ing. Tech. 2016, 88, 1561–1585. [Google Scholar] [CrossRef] [Green Version]
- Feinle, A.; Elsaesser, M.S.; Huesing, N. Sol–gel synthesis of monolithic materials with hierarchical porosity. Chem. Soc. Rev. 2016, 45, 3377–3399. [Google Scholar] [CrossRef]
- Nadargi, D.Y.; Latthe, S.S.; Venkateswara Rao, A. Effect of post-treatment (gel aging) on the properties of methyltrimethoxysilane based silica aerogels prepared by two-step sol–gel process. J. Sol. Gel Sci. Technol. 2009, 49, 53–59. [Google Scholar] [CrossRef]
- Huesing, N.; Schwertfeger, F.; Tappert, W.; Schubert, U. Influence of supercritical drying fluid on structure and properties of organically modified silica aerogels. J. Non-Cryst. Solids 1995, 186, 37–43. [Google Scholar] [CrossRef]
- Brandhuber, D.; Huesing, N.; Raab, C.K.; Torma, V.; Peterlik, H. Cellular mesoscopically organized silica monoliths with tailored surface chemistry by one-step drying/extraction/surface modification processes. J. Mater. Chem. 2005, 15, 1801–1806. [Google Scholar] [CrossRef]
- Estella, J.; Echeverría, J.C.; Laguna, M.; Garrido, J.J. Effects of aging and drying conditions on the structural and textural properties of silica gels. Microporous Mesoporous Mater. 2007, 102, 274–282. [Google Scholar] [CrossRef]
- Aegerter, M.; Leventis, N.; Koebel, M.M. Aerogels Handbook; Springer Science & Business Media: New York, NY, USA, 2011. [Google Scholar]
- Iswar, S.; Malfait, W.J.; Balog, S.; Winnefeld, F.; Lattuda, M.; Koebel, M.M. Effect of aging on silica aerogel properties. Microporous Mesoporous Mater. 2017, 241, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Vareda, J.P.; Matia, T.; Duãres, L. Facile preparation of ambient pressure dried aerogel-like monoliths with reduced shrinkage based on vinyl-modified silica networks. Ceram. Int. 2018, 44, 17453–17458. [Google Scholar] [CrossRef]
- Stojanovic, A.; Comesaña, S.P.; Rentsch, D.; Koebel, M.M.; Malfait, W.J. Ambient pressure drying of silica aerogels after hydrophobization with mono-,di- and tri-functional silanes and mixtures thereof. Microporous Mesoporous Mater. 2019, 284, 289–295. [Google Scholar] [CrossRef]
- Du, D.; Jiang, Y.; Feng, J.; Li, L.; Feng, J. Facile synthesis of silica aerogel composites via ambient-pressure drying without surface modification or solvent exchange. Vacuum 2020, 173, 109117. [Google Scholar] [CrossRef]
- Ding, J.; Zhong, K.; Liu, S.; Wu, X.; Shen, X.; Cui, S.; Chen, X. Flexible and super hydrophobic polymethylsilsesquioxane based silica aerogel for organic solvent adsorption via ambient pressure drying technique. Powder Technol. 2020, 737, 716–726. [Google Scholar] [CrossRef]
- Scherer, G.W. Aging and drying of gels. J. Non Cryst. Solids 1988, 100, 77–92. [Google Scholar] [CrossRef]
- Soleimani Dorcheh, A.; Abbasi, M.H. Silica aerogel; synthesis, properties and characterization. J. Mater. Process. Technol. 2008, 199, 10–26. [Google Scholar] [CrossRef]
- Freytag, A.; Sánchez-Paradinas, S.; Naskar, S.; Wendt, N.; Colombo, M.; Pugliese, G.; Poppe, J.; Demirci, C.; Kretschmer, I.; Bahnemann, D.W.; et al. Versatile Aerogel Fabrication by Freezing and Subsequent Freeze-Drying of Colloidal Nanoparticle Solutions. Angew. Chem. Int. Ed. 2016, 55, 1200–1203. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; He, S.; Gong, L.; Cheng, X.; Li, C.; Li, Z.; Liu, Z.; Zhang, H. Low thermal-conductivity and high thermal stable silica aerogel based on MTMS/Water-glass co-precursor prepared by freeze drying. Mater. Des. 2017, 113, 246–253. [Google Scholar] [CrossRef]
- Kistler, S.S. Coherent Expanded-Aerogels. J. Phys. Chem. 1932, 36, 52–64. [Google Scholar] [CrossRef]
- Van Bommel, M.J.; de Haan, A.B. Drying of silica aerogel with supercritical carbon dioxide. J. Non. Cryst. Solids 1995, 186, 78–82. [Google Scholar] [CrossRef]
- Riffat, S.B.; Qiu, G. A review of state-of-the-art aerogel applications in buildings. Int. J. Low Carbon Technol. 2013, 8, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Boukind, S.; Sair, S.; Ousaleh, H.A.; Mansouri, S.; Zahouily, M.; Abboud, Y.; El Bouari, A. Ambient Pressure Drying as an Advanced Approach to the Synthesis of Silica Aerogel Composite for Building Thermal Insulation. J. Nat. Fibers 2021, 19, 10142–10156. [Google Scholar] [CrossRef]
- Impéror-Clerc, M.; Davidson, P.; Davidson, A. Existence of a Microporous Corona around the Mesopores of Silica-Based SBA-15 Materials Templated by Triblock Copolymers. J. Am. Chem. Soc. 2000, 122, 11925–11933. [Google Scholar] [CrossRef]
- Rao, A.P.; Rao, A.V.; Pajonk, G.M. Hydrophobic and physical properties of the ambient pressure dried silica aerogels with sodium silicate precursor using various surface modification agents. Appl. Surf. Sci. 2007, 253, 6032–6040. [Google Scholar] [CrossRef]
- Scherer, G.W. Theory of Drying. J. Am. Ceram. Soc. 1990, 73, 3–14. [Google Scholar] [CrossRef]
- Galarneau, A.; Cambon, H.; Di Renzo, F.; Ryoo, R.; Choi, M.; Fajula, F. Microporosity and connections between pores in SBA-15 mesostructured silicas as a function of the temperature of synthesis. New J. Chem. 2003, 27, 73–79. [Google Scholar] [CrossRef]
- Joo, S.H.; Ryoo, R.; Kruk, M.; Jaroniec, M. Evidence for General Nature of Pore Interconnectivity in 2-Dimensional Hexagonal Mesoporous Silicas Prepared Using Block Copolymer Templates. J. Phys. Chem. B 2002, 106, 4640–4646. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M.; Ko, C.H.; Ryoo, R. Characterization of the Porous Structure of SBA-15. Chem. Mater. 2000, 12, 1961–1968. [Google Scholar] [CrossRef]
- Brinker, C.J.; Scherer, G.W. Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing; Academic Press: New York, NY, USA, 1990. [Google Scholar]
- Gross, J.; Scherer, G.W. Dynamic pressurization: Novel method for measuring fluid permeability. J. Non Cryst. Solids 2003, 325, 34–47. [Google Scholar] [CrossRef]
- Reichenauer, G. Thermal aging of silica gels in water. J. Non Cryst. Solids 2004, 350, 189–195. [Google Scholar] [CrossRef]
- Balzer, C.; Waag, A.M.; Putz, F.; Huesing, N.; Paris, O.; Gor, G.Y.; Neimark, A.V.; Reichenauer, G. Mechanical Characterization of Hierarchical Structured Porous Silica by in situ Dilatometry Measurements during Gas Adsorption. Langmuir 2019, 35, 2948–2956. [Google Scholar] [CrossRef]
- Balzer, C. Adsorption-Induced Deformation of Nanoporous Materials–In-Situ Dilatometry and Modelin. Ph.D. Thesis, Physics Department, Wuerzburg University, Würzburg, Germany, 2017. [Google Scholar]
- Kistler, S.S. Coherent Expanded Aerogels and Jellies. Nature 1931, 127, 741. [Google Scholar] [CrossRef]
- Prakash, S.S.; Brinker, C.J.; Hurd, A.J. Silica aerogel films at ambient pressure. J. Non Cryst. Solids 1995, 190, 264–275. [Google Scholar] [CrossRef]
- Von der Lehr, M.; Hormann, K.; Höltzel, A.; White, L.S.; Reising, A.E.; Bertino, M.F.; Smarsly, B.; Tallarek, U. Mesopore etching under supercritical conditions–A shortcut to hierarchically porous silica monoliths. Microporous Mesoporous Mater. 2017, 243, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Putz, F.; Waag, A.; Balzer, C.; Braxmeier, S.; Elsaesser, M.S.; Ludescher, L.; Paris, O.; Malfait, W.J.; Reichenauer, G.; Huesing, N. The influence of drying and calcination on the surface chemistry, pore structure and mechanical properties of hierarchically organized porous silica monoliths. Microporous Mesoporous Mater. 2019, 288, 109578. [Google Scholar] [CrossRef]
- Woigner, T.; Phalippou, J.; Quinson, J.F.; Pauthe, M.; Laveissiere, F. Physicochemical transformation of silica gels during hypercritical drying. J. Non Cryst. Solids 1992, 145, 25–32. [Google Scholar] [CrossRef]
- van Oss, C.J. Chapter Two–The Apolar and Polar Properties of Liquid Water and Other Condensed-Phase Materials. Interface Sci. Technol. 2008, 16, 13–30. [Google Scholar] [CrossRef]
- Schwan, M.; Nefzger, S.; Zoghi, B.; Oligschleger, C.; Milow, B. Improvement of Solvent Exchange for Supercritical Dried Aerogels. Front. Mater. 2021, 8, 662487. [Google Scholar] [CrossRef]
- Scherer, G.W. Recent progress in drying of gels. J. Non Cryst. Solids 1992, 147, 363–374. [Google Scholar] [CrossRef]
- Scherer, G.W. Cavitation during drying of a gel. J. Non Cryst. Solids 1995, 189, 197–211. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Hishikura, H.; Iura, J.; Kokubu, Y. Monolithic dried gels and silica glass prepared by the sol–gel process. J. Non Cryst. Solids 1984, 63, 61–69. [Google Scholar] [CrossRef]
- Reichenauer, G.; Pfrang, T.; Hofmann, M. Drying of meso- and macroporous gels–Length change and drying dynamics. Colloids Surf. A: Physicochem. Eng. Asp. 2007, 300, 211–215. [Google Scholar] [CrossRef]
- Fricke, J.; Emmerling, A. Aerogels–Preparation, properties, applications. In Structure and Bonding 77; Springer: Berlin, Germany, 1992; pp. 37–87. [Google Scholar]
- Mulder, C.A.M.; van Lierop, J.G. Preparation, Densification and Characterization of Autoclave dried SiO2 Gels. In Aerogels; Fricke, J., Ed.; Springer: Berlin, Germany, 1986; pp. 68–75. [Google Scholar]
- Glatter, O.; Kratky, O. Small Angle X-ray Scattering; Academic Press: London, UK, 1982. [Google Scholar]
- Brandhuber, D.; Torma, V.; Raab, C.; Peterlik, H.; Kulak, A.; Huesing, N. Glycol-Modified Silanes in the Synthesis of Mesoscopically Organized Silica Monoliths with Hierarchical Porosity. Chem. Mater. 2005, 17, 4262–4271. [Google Scholar] [CrossRef]
- Koczwara, C.; Rumswinkel, S.; Hammerschmidt, L.; Salihovic, M.; Elsaesser, M.S.; Amenitsch, H.; Paris, O.; Huesing, N. Nanofibers versus Nanopores: A Comparison of the Electrochemical Performance of Hierarchically Ordered Porous Carbons. ACS Appl. Energy Mater. 2019, 2, 5279–5291. [Google Scholar] [CrossRef]
- Selmer, I.; Behnecke, A.-S.; Quiño, J.; Braeuer, A.S.; Gurikov, P.; Smirnova, I. Model development for sc-drying kinetics of aerogels: Part Monoliths and single particles. J. Supercrit. Fluids 2018, 140, 415–430. [Google Scholar] [CrossRef]
- Bueno, A.; Selmer, I.; Raman, S.P.; Gurikov, P.; Lölsberg, W.; Weinrich, D.; Fricke, M.; Smirnova, I. First Evidence of Solvent Spillage under Subcritical Conditions in Aerogel Production. Ind. Eng. Chem. Res. 2018, 57, 8698–8707. [Google Scholar] [CrossRef]
- Kohns, R.; Anders, N.; Enke, D.; Tallarek, U. Influence of Pore Space Hierarchy on the Efficiency of an Acetylcholinesterase-Based Support for Biosensorics. Adv. Mat. Interfaces 2021, 8, 2000163. [Google Scholar] [CrossRef]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the BET equation applicable to microporous adsorbents? Stud. Surf. Sci. Catal. 2007, 160, 49–56. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ρbulk/g cm–3 | ρskela/g cm–3 | Φb/% | SBETc/m2 g−1 | Vmicroc/cm3 g−1 | Vmeso,DFTc/cm3 g−1 | Vmacrod/cm3 g−1 | dmeso, DFTc/nm | ae/nm |
|---|---|---|---|---|---|---|---|---|---|
| SCD | 0.252 ± 0.006 | 1.73 ± 0.05 | 85 ± 0.4 | 1015 ± 78 | 0.06 ± 0.00 | 1.06 ± 0.12 | 1.72 | 6.6 ± 0.12 | 11.4 |
| 86 °C | 0.272 ± 0.006 | 1.66 ± 0.06 | 84 ± 0.3 | 1277 ± 34 | 0.11 ± 0.00 | 1.19 ± 0.05 | 1.97 | 6.8 ± 0.00 | n.d. |
| 82 °C | 0.262 ± 0.008 | 1.67 ± 0.06 | 84 ± 0.5 | 1297 ± 17 | 0.10 ± 0.01 | 1.24 ± 0.09 | 2.11 | 6.9 ± 0.19 | n.d. |
| 78 °C | 0.232 ± 0.003 | 1.70 ± 0.05 | 86 ± 0.2 | 1622 ± 45 | 0.04 ± 0.03 | 1.84 ± 0.12 | 2.31 | 8.5 ± 0.12 | 13.0 |
| 74 °C | 0.225 ± 0.005 | 1.72 ± 0.05 | 87 ± 0.3 | 1616 ± 68 | 0.01 ± 0.00 | 1.91 ± 0.09 | 2.51 | 8.7 ± 0.08 | 12.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kohns, R.; Torres-Rodríguez, J.; Euchler, D.; Seyffertitz, M.; Paris, O.; Reichenauer, G.; Enke, D.; Huesing, N. Drying of Hierarchically Organized Porous Silica Monoliths–Comparison of Evaporative and Supercritical Drying. Gels 2023, 9, 71. https://doi.org/10.3390/gels9010071
Kohns R, Torres-Rodríguez J, Euchler D, Seyffertitz M, Paris O, Reichenauer G, Enke D, Huesing N. Drying of Hierarchically Organized Porous Silica Monoliths–Comparison of Evaporative and Supercritical Drying. Gels. 2023; 9(1):71. https://doi.org/10.3390/gels9010071
Chicago/Turabian StyleKohns, Richard, Jorge Torres-Rodríguez, Daniel Euchler, Malina Seyffertitz, Oskar Paris, Gudrun Reichenauer, Dirk Enke, and Nicola Huesing. 2023. "Drying of Hierarchically Organized Porous Silica Monoliths–Comparison of Evaporative and Supercritical Drying" Gels 9, no. 1: 71. https://doi.org/10.3390/gels9010071