
The Genome-Wide Characterization of Alternative Splicing and RNA Editing in the Development of Coprinopsis cinerea

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Cultivation, Sample Collection, and High-Throughput Sequencing
2.2. Read Mapping and Counting
2.3. Identification of Alternative Splicing
2.4. Identification of RNA Editing
2.5. Reverse Transcription and PCR Amplification
2.6. Functional Annotations and Analyses
3. Results and Discussion
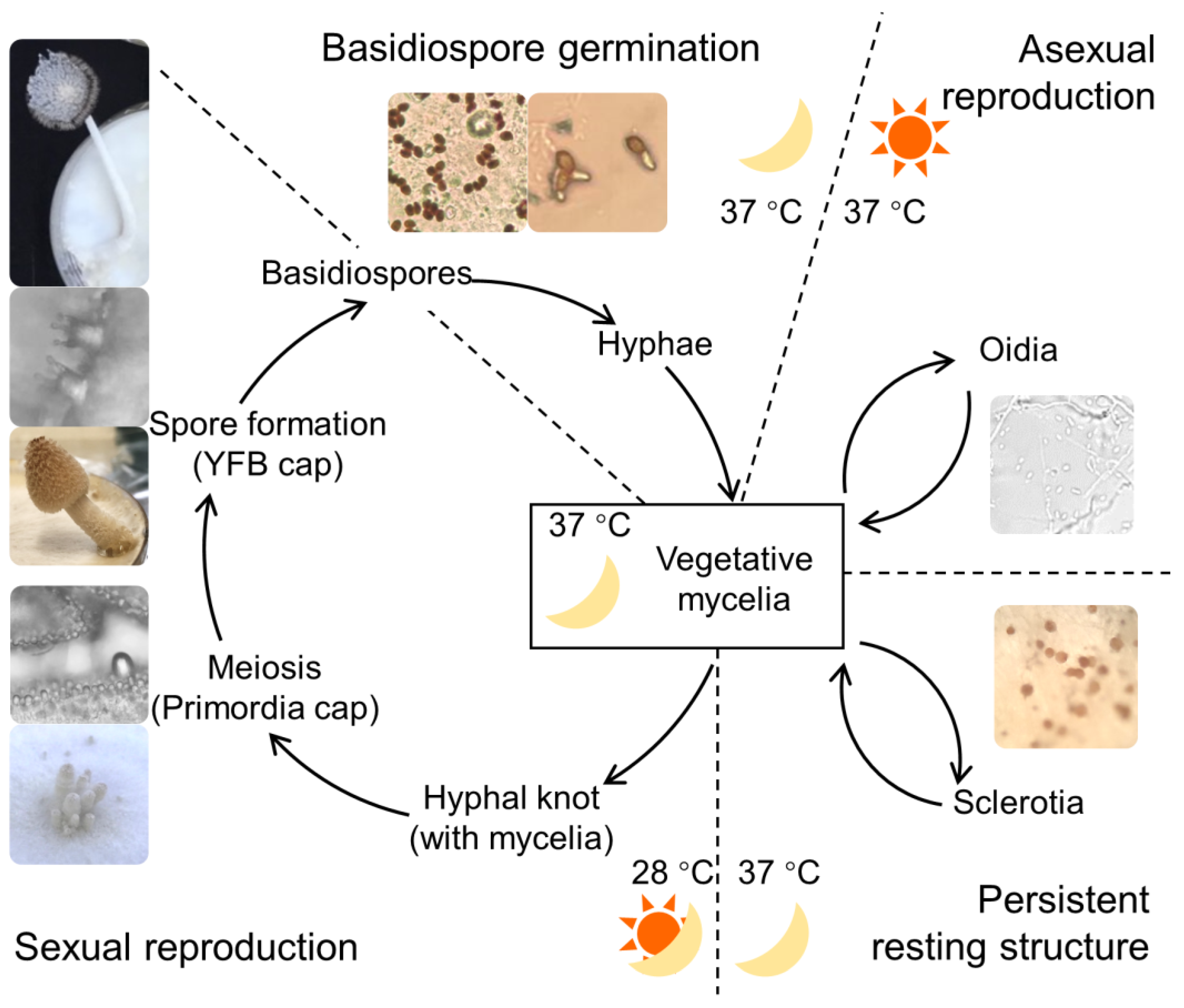
3.1. Overview of C. cinerea Developmental Transitions
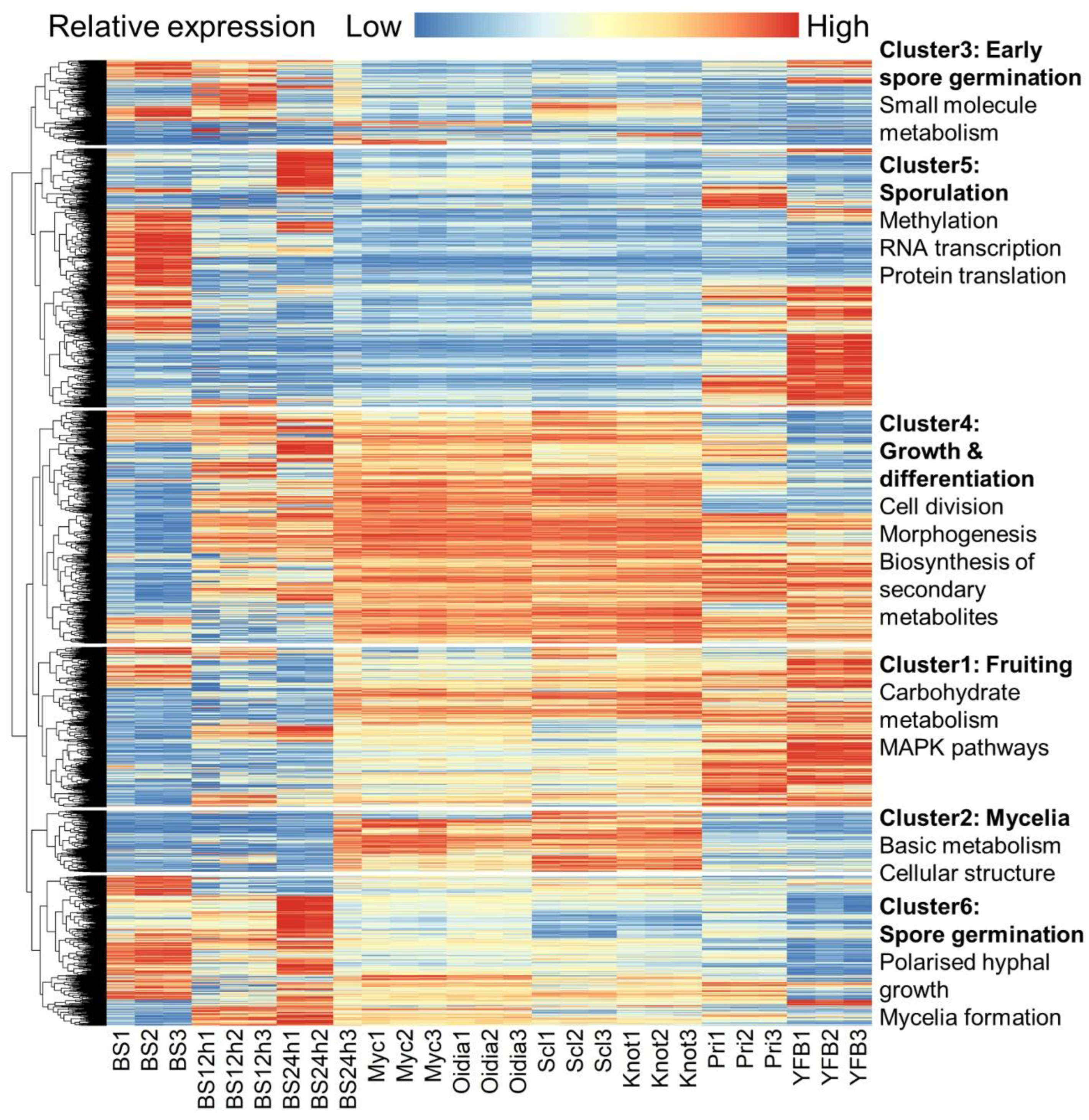
3.2. Gene Expression Profiles in Different Developmental Trajectories
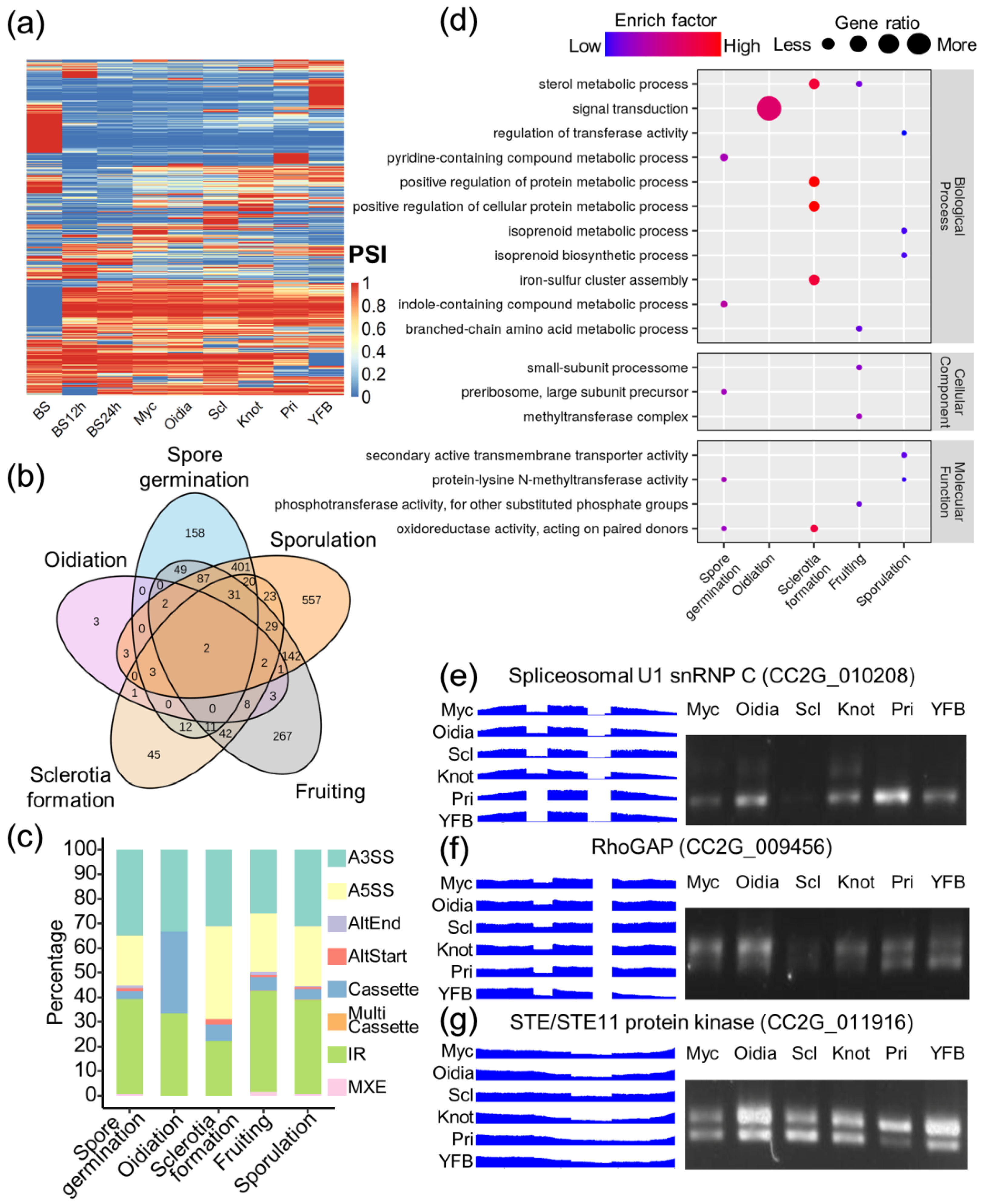
3.3. Alternative Splicing Landscape in C. cinerea
3.4. Alternative Splicing Generated Stage-Specific Isoforms during Development
3.5. Broad Effects of Developmentally Regulated Alternative Splicing in C. cinerea
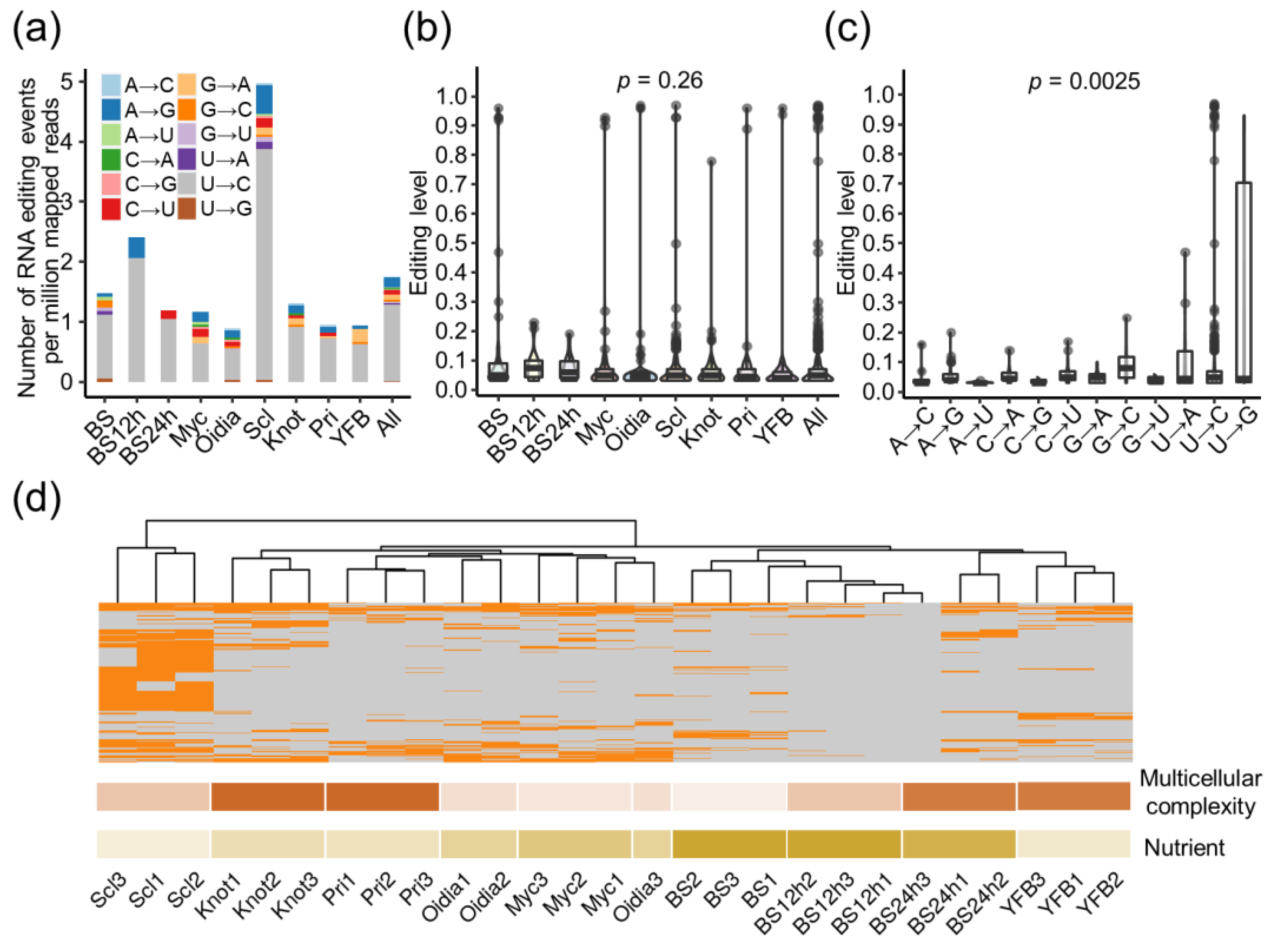
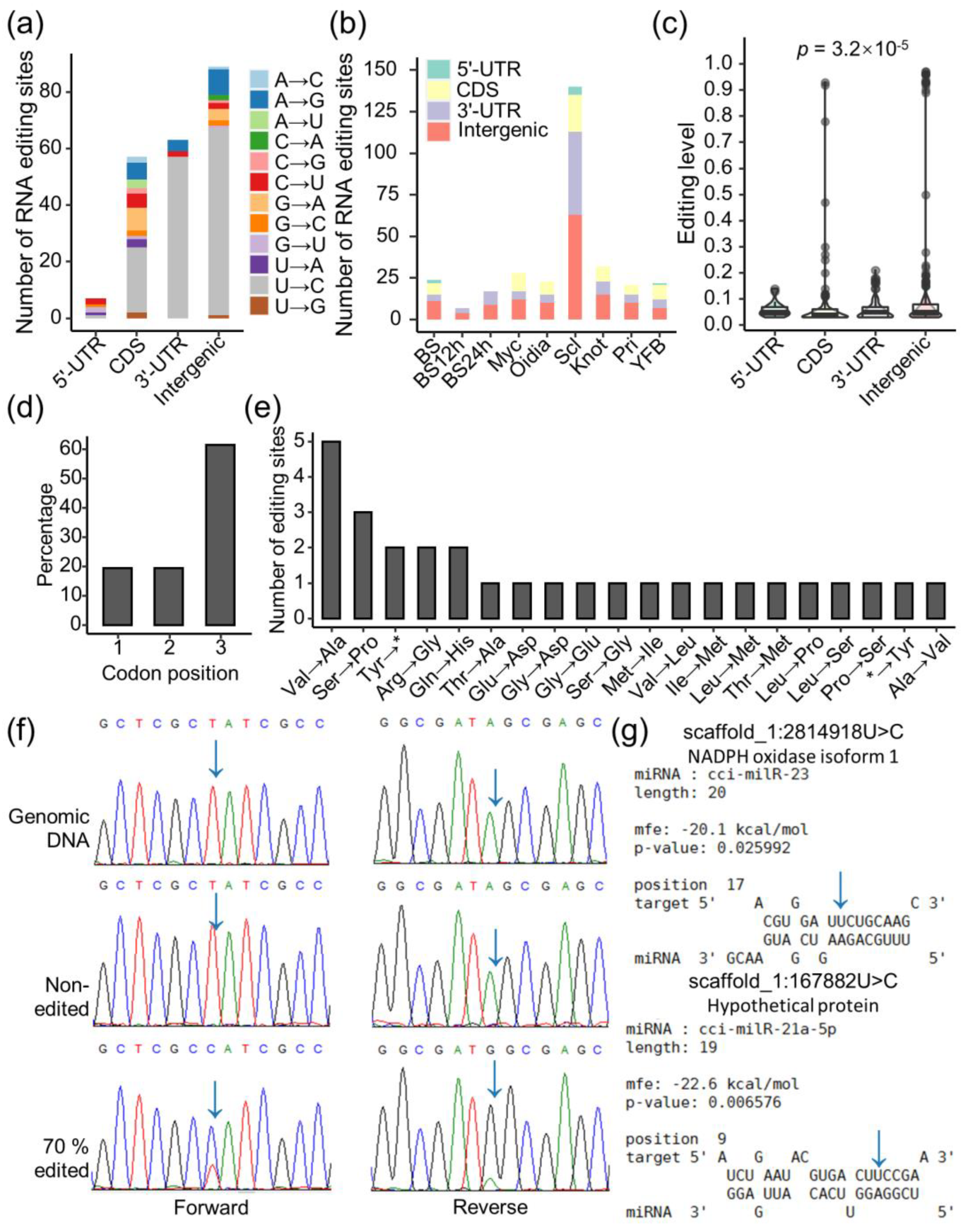
3.6. RNA Editing Happened in a Stage-Specific Manner along the Development of C. cinerea
3.7. RNA Editing Diversifies the Regulation of Gene Expression
3.8. Putative Catalytic Genes of RNA Editing in C. cinerea
3.9. Alternative Splicing and RNA Editing Together Provided Adaptations in Development
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tedersoo, L.; Sánchez-Ramírez, S.; Kõljalg, U.; Bahram, M.; Döring, M.; Schigel, D.; May, T.; Ryberg, M.; Abarenkov, K. High-Level Classification of the Fungi and a Tool for Evolutionary Ecological Analyses. Fungal Divers. 2018, 90, 135–159. [Google Scholar] [CrossRef]
- He, M.-Q.; Zhao, R.-L.; Liu, D.-M.; Denchev, T.T.; Begerow, D.; Yurkov, A.; Kemler, M.; Millanes, A.M.; Wedin, M.; McTaggart, A.R.; et al. Species Diversity of Basidiomycota. Fungal Divers. 2022, 114, 281–325. [Google Scholar] [CrossRef]
- Kües, U. Life History and Developmental Processes in the Basidiomycete Coprinus cinereus. Microbiol. Mol. Biol. Rev. 2000, 64, 316–353. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Haelewaters, D.; Schoutteten, N.; Begerow, D.; Boekhout, T.; Giachini, A.J.; Gorjón, S.P.; Gunde-Cimerman, N.; Hyde, K.D.; Kemler, M.; et al. Delimiting Species in Basidiomycota: A Review. Fungal Divers. 2021, 109, 181–237. [Google Scholar] [CrossRef]
- Virágh, M.; Merényi, Z.; Csernetics, Á.; Földi, C.; Sahu, N.; Liu, X.-B.; Hibbett, D.S.; Nagy, L.G. Evolutionary Morphogenesis of Sexual Fruiting Bodies in Basidiomycota: Toward a New Evo-Devo Synthesis. Microbiol. Mol. Biol. Rev. 2022, 86, e00019-21. [Google Scholar] [CrossRef] [PubMed]
- Kues, U.; Liu, Y. Fruiting Body Production in Basidiomycetes. Appl. Microbiol. Biotechnol. 2000, 54, 141–152. [Google Scholar] [CrossRef]
- Xie, Y.; Chang, J.; Kwan, H.S. Carbon Metabolism and Transcriptome in Developmental Paths Differentiation of a Homokaryotic Coprinopsis cinerea Strain. Fungal Genet. Biol. 2020, 143, 103432. [Google Scholar] [CrossRef]
- Sakamoto, Y. Influences of Environmental Factors on Fruiting Body Induction, Development and Maturation in Mushroom-Forming Fungi. Fungal Biol. Rev. 2018, 32, 236–248. [Google Scholar] [CrossRef]
- Srivilai, P.; Chaisaena, W.; Kües, U. Genetic Analysis of Coprinopsis cinerea Mutants with Defects in Fruiting Body Development. In VI Genetics and Cellular Biology of Basidiomycetes; Antonio, G.P., Lucy, R., Eds.; Universida Pública de Navarra: Pamplona, Spain, 2005; pp. 177–190. ISBN 8497691075. [Google Scholar]
- Swamy, S.; Uno, I.; Ishikawa, T. Morphogenetic Effects of Mutations at the A and B Incompatibility Factors in Coprinus cinereus. J. Gen. Microbiol. 1984, 130, 3219–3224. [Google Scholar] [CrossRef]
- Xie, Y.; Zhong, Y.; Chang, J.; Kwan, H.S. Chromosome-Level de Novo Assembly of Coprinopsis cinerea A43mut B43mut Pab1-1 #326 and Genetic Variant Identification of Mutants Using Nanopore MinION Sequencing. Fungal Genet. Biol. 2021, 146, 103485. [Google Scholar]
- Kamada, T.; Sano, H.; Nakazawa, T.; Nakahori, K. Regulation of Fruiting Body Photomorphogenesis in Coprinopsis cinerea. Fungal Genet. Biol. 2010, 47, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Moore, D. Developmental Genetics of Coprinus cinereus: Genetic Evidence That Carpophores and Sclerotia Share a Common Pathway of Initiation. Curr. Genet. 1981, 3, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Cramer, P. Organization and Regulation of Gene Transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Schaefke, B.; Sun, W.; Li, Y.S.; Fang, L.; Chen, W. The Evolution of Posttranscriptional Regulation. Wiley Interdiscip. Rev. RNA 2018, 9, e1485. [Google Scholar] [CrossRef] [PubMed]
- Kües, U. From Two to Many: Multiple Mating Types in Basidiomycetes. Fungal Biol. Rev. 2015, 29, 126–166. [Google Scholar] [CrossRef]
- Liu, C.; Kang, L.; Lin, M.; Bi, J.; Liu, Z.; Yuan, S. Molecular Mechanism by Which the GATA Transcription Factor CcNsdD2 Regulates the Developmental Fate of Coprinopsis cinerea under Dark or Light Conditions. MBio 2022, 13, e03626-21. [Google Scholar] [CrossRef]
- Chavez, L.; Huang, Y.; Luong, K.; Agarwal, S.; Iyer, L.M.; Pastor, W.A.; Hench, V.K.; Frazier-Bowers, S.A.; Korol, E.; Liu, S.; et al. Simultaneous Sequencing of Oxidized Methylcytosines Produced by TET/JBP Dioxygenases in Coprinopsis cinerea. Proc. Natl. Acad. Sci. USA 2014, 111, E5149–E5158. [Google Scholar] [CrossRef]
- Chang, S.S.; Zhang, Z.; Liu, Y. RNA Interference Pathways in Fungi: Mechanisms and Functions. Annu. Rev. Microbiol. 2012, 66, 305–323. [Google Scholar] [CrossRef]
- Wang, M.; Dean, R.A. Movement of Small RNAs in and between Plants and Fungi. Mol. Plant Pathol. 2020, 21, 589–601. [Google Scholar] [CrossRef]
- Lau, A.Y.T.; Cheng, X.; Cheng, C.K.; Nong, W.; Cheung, M.K.; Chan, R.H.-F.; Hui, J.H.L.; Kwan, H.S. Discovery of MicroRNA-like RNAs during Early Fruiting Body Development in the Model Mushroom Coprinopsis cinerea. PLoS ONE 2018, 13, e0198234. [Google Scholar] [CrossRef]
- Lau, A.Y.T.; Xie, Y.; Cheung, M.K.; Cheung, P.C.K.; Kwan, H.S. Genome-Wide mRNA and miRNA Analysis in the Early Stages of Germ Tube Outgrowth in Coprinopsis cinerea. Fungal Genet. Biol. 2020, 142, 103416. [Google Scholar] [CrossRef]
- Ule, J.; Blencowe, B.J. Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol. Cell 2019, 76, 329–345. [Google Scholar] [CrossRef]
- Chaudhary, S.; Khokhar, W.; Jabre, I.; Reddy, A.S.N.; Byrne, L.J.; Wilson, C.M.; Syed, N.H. Alternative Splicing and Protein Diversity: Plants versus Animals. Front. Plant Sci. 2019, 10, 708. [Google Scholar] [CrossRef]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic Atlas of Mushroom Development Reveals Conserved Genes behind Complex Multicellularity in Fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Chen, D.; Qi, Z.; Wang, H.; Chen, Y.; Wang, Q.; Jiang, C.; Xu, J.R.; Liu, H. Landscape and Regulation of Alternative Splicing and Alternative Polyadenylation in a Plant Pathogenic Fungus. New Phytol. 2022, 235, 674–689. [Google Scholar] [CrossRef]
- Gray, M.W. Evolutionary Origin of RNA Editing. Biochemistry 2012, 51, 5235–5242. [Google Scholar] [CrossRef] [PubMed]
- Liscovitch-Brauer, N.; Alon, S.; Porath, H.T.; Elstein, B.; Unger, R.; Ziv, T.; Admon, A.; Levanon, E.Y.; Rosenthal, J.J.C.; Eisenberg, E. Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 2017, 169, 191–202. [Google Scholar] [CrossRef]
- Gerber, A.P.; Keller, W. RNA Editing by Base Deamination: More Enzymes, More Targets, New Mysteries. Trends Biochem. Sci. 2001, 26, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Licht, K.; Jantsch, M.F. Rapid and Dynamic Transcriptome Regulation by RNA Editing and RNA Modifications. J. Cell Biol. 2016, 213, 15–22. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA Editing—Immune Protector and Transcriptome Diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Ichinose, M.; Sugita, M. RNA Editing and Its Molecular Mechanism in Plant Organelles. Genes 2017, 8, 5. [Google Scholar] [CrossRef]
- Zhu, Y.; Luo, H.; Zhang, X.; Song, J.; Sun, C.; Ji, A.; Xu, J.; Chen, S. Abundant and Selective RNA-Editing Events in the Medicinal Mushroom Ganoderma lucidum. Genetics 2014, 196, 1047–1057. [Google Scholar] [CrossRef]
- Liu, H.; Li, Y.; Chen, D.; Qi, Z.; Wang, Q.; Wang, J.; Jiang, C.; Xu, J.-R. A-to-I RNA Editing Is Developmentally Regulated and Generally Adaptive for Sexual Reproduction in Neurospora crassa. Proc. Natl. Acad. Sci. USA 2017, 114, E7756–E7765. [Google Scholar] [CrossRef]
- Liu, H.; Wang, Q.; He, Y.; Chen, L.; Hao, C.; Jiang, C.; Li, Y.; Dai, Y.; Kang, Z.; Xu, J.R. Genome-Wide A-to-I RNA Editing in Fungi Independent of ADAR Enzymes. Genome Res. 2016, 26, 499–509. [Google Scholar] [CrossRef]
- Xin, K.; Zhang, Y.; Fan, L.; Qi, Z.; Feng, C.; Wang, Q.; Jiang, C.; Xu, J.-R.; Liu, H. Experimental Evidence for the Functional Importance and Adaptive Advantage of A-to-I RNA Editing in Fungi. Proc. Natl. Acad. Sci. USA 2023, 120, e2219029120. [Google Scholar] [CrossRef] [PubMed]
- Nowrousian, M. Genomics and Transcriptomics to Study Fruiting Body Development: An Update. Fungal Biol. Rev. 2018, 32, 231–235. [Google Scholar] [CrossRef]
- Merényi, Z.; Virágh, M.; Gluck-Thaler, E.; Slot, J.C.; Kiss, B.; Varga, T.; Geösel, A.; Hegedüs, B.; Bálint, B.; Nagy, L.G. Gene Age Shapes the Transcriptional Landscape of Sexual Morphogenesis in Mushroom-Forming Fungi (Agaricomycetes). Elife 2022, 11, e71348. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Wu, B.; Gaskell, J.; Zhang, J.; Toapanta, C.; Ahrendt, S.; Blanchette, R.A.; Master, E.; Cullen, D.; Hibbett, D.S.; et al. RNA-Editing in Basidiomycota, Revisited. ISME Commun. 2021, 1, 70. [Google Scholar] [CrossRef]
- Muraguchi, H.; Umezawa, K.; Niikura, M.; Yoshida, M.; Kozaki, T.; Ishii, K.; Sakai, K.; Shimizu, M.; Nakahori, K.; Sakamoto, Y.; et al. Strand-Specific RNA-Seq Analyses of Fruiting Body Development in Coprinopsis cinerea. PLoS ONE 2015, 10, e0141586. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome Assembly from Long-Read RNA-Seq Alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zong, J.; Wei, N.; Cheng, J.; Zhou, X.; Cheng, Y.; Chen, D.; Guo, Q.; Zhang, B.; Feng, Y. CASH: A Constructing Comprehensive Splice Site Method for Detecting Alternative Splicing Events. Brief. Bioinform. 2018, 19, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.T.; Knop, K.; Barton, G.J.; Simpson, G.G. 2passtools: Two-Pass Alignment Using Machine-Learning-Filtered Splice Junctions Increases the Accuracy of Intron Detection in Long-Read RNA Sequencing. Genome Biol. 2021, 22, 72. [Google Scholar] [CrossRef]
- Gatto, A.; Torroja-Fungairiño, C.; Mazzarotto, F.; Cook, S.A.; Barton, P.J.R.; Sánchez-Cabo, F.; Lara-Pezzi, E. FineSplice, Enhanced Splice Junction Detection and Quantification: A Novel Pipeline Based on the Assessment of Diverse RNA-Seq Alignment Solutions. Nucleic Acids Res. 2014, 42, e71. [Google Scholar] [CrossRef]
- Picardi, E.; Pesole, G. REDItools: High-Throughput RNA Editing Detection Made Easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA Target Prediction Easy, Fast and Flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kwan, H.S.; Chan, P.L.; Wu, W.J.; Chiou, J.; Chang, J. The Phylotranscriptomic Hourglass Pattern in Fungi: An Updated Model. bioRxiv 2022. bioRxiv:2022.07.14.500038. [Google Scholar] [CrossRef]
- Cheng, X.; Hui, J.H.L.; Lee, Y.Y.; Law, P.T.W.; Kwan, H.S. A “Developmental Hourglass” in Fungi. Mol. Biol. Evol. 2015, 32, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps, version 1.0.12. 2019.
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2022. [Google Scholar]
- Grützmann, K.; Szafranski, K.; Pohl, M.; Voigt, K.; Petzold, A.; Schuster, S. Fungal Alternative Splicing Is Associated with Multicellular Complexity and Virulence: A Genome-Wide Multi-Species Study. DNA Res. 2014, 21, 27–39. [Google Scholar] [CrossRef]
- Fang, S.; Hou, X.; Qiu, K.; He, R.; Feng, X.; Liang, X. The Occurrence and Function of Alternative Splicing in Fungi. Fungal Biol. Rev. 2020, 34, 178–188. [Google Scholar] [CrossRef]
- García-Moreno, J.F.; Romão, L. Perspective in Alternative Splicing Coupled to Nonsense-Mediated mRNA Decay. Int. J. Mol. Sci. 2020, 21, 9424. [Google Scholar] [CrossRef]
- Han, S.; Kim, D.; Shivakumar, M.; Lee, Y.J.; Garg, T.; Miller, J.E.; Kim, J.H.; Kim, D.; Lee, Y. The Effects of Alternative Splicing on MiRNA Binding Sites in Bladder Cancer. PLoS ONE 2018, 13, e0190708. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, T.; Pelkmans, J.F.; Lugones, L.G.; Wösten, H.A.B.; Abeel, T.; Reinders, M.J.T. Schizophyllum commune Has an Extensive and Functional Alternative Splicing Repertoire. Sci. Rep. 2016, 6, 33640. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, B.; Yin, C.; Guo, Y.; Lin, Y.; Pan, L.; Wang, B. Characterization of Natural Antisense Transcript, Sclerotia Development and Secondary Metabolism by Strand-Specific RNA Sequencing of Aspergillus flavus. PLoS ONE 2014, 9, e97814. [Google Scholar] [CrossRef]
- de Freitas Pereira, M.; Narvaes da Rocha Campos, A.; Anastacio, T.C.; Morin, E.; Brommonschenkel, S.H.; Martin, F.; Kohler, A.; Costa, M.D. The Transcriptional Landscape of Basidiosporogenesis in Mature Pisolithus microcarpus Basidiocarp. BMC Genom. 2017, 18, 157. [Google Scholar] [CrossRef]
- Wang, F.; Sethiya, P.; Hu, X.; Guo, S.; Chen, Y.; Li, A.; Tan, K.; Wong, K.H. Transcription in Fungal Conidia before Dormancy Produces Phenotypically Variable Conidia That Maximize Survival in Different Environments. Nat. Microbiol. 2021, 6, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, R.; Bottardi, S.; Cogoi, S.; Apezteguia, I.; Santoro, C. Alternative Translation Initiation Site Usage Results in Two Functionally Distinct Forms of the GATA-1 Transcription Factor. Proc. Natl. Acad. Sci. USA 1995, 92, 11598–11602. [Google Scholar] [CrossRef] [PubMed]
- Ilouz, R.; Lev-Ram, V.; Bushong, E.A.; Stiles, T.L.; Friedmann-Morvinski, D.; Douglas, C.; Goldberg, G.; Ellisman, M.H.; Taylor, S.S. Isoform-Specific Subcellular Localization and Function of Protein Kinase a Identified by Mosaic Imaging of Mouse Brain. Elife 2017, 6, e17681. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Brenner, S.E. Regulation of Splicing Factors by Alternative Splicing and NMD Is Conserved between Kingdoms yet Evolutionarily Flexible. Mol. Biol. Evol. 2015, 32, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Seo, P.J.; Park, M.J.; Park, C.M. Alternative Splicing of Transcription Factors in Plant Responses to Low Temperature Stress: Mechanisms and Functions. Planta 2013, 237, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Liew, Y.J.; Li, Y.; Baumgarten, S.; Voolstra, C.R.; Aranda, M. Condition-Specific RNA Editing in the Coral Symbiont Symbiodinium microadriaticum. PLoS Genet. 2017, 13, e1006619. [Google Scholar] [CrossRef] [PubMed]
- Farajollahi, S.; Maas, S. Molecular Diversity through RNA Editing: A Balancing Act. Trends Genet. 2010, 26, 221–230. [Google Scholar] [CrossRef]
- Bian, Z.; Ni, Y.; Xu, J.R.; Liu, H. A-to-I mRNA Editing in Fungi: Occurrence, Function, and Evolution. Cell. Mol. Life Sci. 2019, 76, 329–340. [Google Scholar] [CrossRef]
- Duan, Y.; Li, H.; Cai, W. Adaptation of A-to-I RNA Editing in Bacteria, Fungi, and Animals. Front. Microbiol. 2023, 14, 1643. [Google Scholar] [CrossRef]
- Ruchika; Okudaira, C.; Sakari, M.; Tsukahara, T. Genome-Wide Identification of U-To-C RNA Editing Events for Nuclear Genes in Arabidopsis thaliana. Cells 2021, 10, 635. [Google Scholar] [CrossRef]
- Knoop, V. C-to-U and U-to-C: RNA Editing in Plant Organelles and Beyond. J. Exp. Bot. 2023, 74, 2273–2294. [Google Scholar] [CrossRef] [PubMed]
- Ruchika; Tsukahara, T. The U-to-C RNA Editing Affects the mRNA Stability of Nuclear Genes in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2021, 571, 110–117. [Google Scholar] [CrossRef]
- Takenaka, M.; Takenaka, S.; Barthel, T.; Frink, B.; Haag, S.; Verbitskiy, D.; Oldenkott, B.; Schallenberg-Rüdinger, M.; Feiler, C.G.; Weiss, M.S.; et al. DYW Domain Structures Imply an Unusual Regulation Principle in Plant Organellar RNA Editing Catalysis. Nat. Catal. 2021, 4, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Kawabata, M.; Akaiwa, Y.; Shimajiri, Y.; Nakamura, I.; Tamai, T.; Nakamura, T.; Yagi, Y.; Gutmann, B. U-to-C RNA Editing by Synthetic PPR-DYW Proteins in Bacteria and Human Culture Cells. Commun. Biol. 2022, 5, 968. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J. Evolution of Sexual Reproduction: A View from the Fungal Kingdom Supports an Evolutionary Epoch with Sex before Sexes. Fungal Biol. Rev. 2015, 29, 108–117. [Google Scholar] [CrossRef]
- Wallen, R.M.; Perlin, M.H. An Overview of the Function and Maintenance of Sexual Reproduction in Dikaryotic Fungi. Front. Microbiol. 2018, 9, 503. [Google Scholar] [CrossRef] [PubMed]
- Coley-Smith, J.R.; Cooke, R.C. Survival and Germination of Fungal Sclerotia. Annu. Rev. Phytopathol. 1971, 9, 65–92. [Google Scholar] [CrossRef]
- Alon, S.; Garrett, S.C.; Levanon, E.Y.; Olson, S.; Graveley, B.R.; Rosenthal, J.J.C.; Eisenberg, E. The Majority of Transcripts in the Squid Nervous System Are Extensively Recoded by A-to-I RNA Editing. Elife 2015, 4, e05198. [Google Scholar] [CrossRef]
- Laloum, T.; Martín, G.; Duque, P. Alternative Splicing Control of Abiotic Stress Responses. Trends Plant Sci. 2018, 23, 140–150. [Google Scholar] [CrossRef]
- Hsiao, Y.-H.E.; Bahn, J.H.; Yang, Y.; Lin, X.; Tran, S.; Yang, E.-W.; Quinones-Valdez, G.; Xiao, X. RNA Editing in Nascent RNA Affects Pre-mRNA Splicing. Genome Res. 2018, 28, 812–823. [Google Scholar] [CrossRef]
- Polak, E.; Hermann, R.; Kües, U.; Aebi, M. Asexual Sporulation in Coprinus cinereus: Structure and Development of Oidiophores and Oidia in an Amut Bmut Homokaryon. Fungal Genet. Biol. 1997, 22, 112–126. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage (abb.) | Incubation |
|---|---|
| Vegetative mycelia (Myc) | Continuous darkness at 37 °C for 4 d |
| Oidia-forming mycelia (Oidia) | Continuous light at 37 °C for 4 d |
| Sclerotia-forming mycelia (Scl) | Continuous darkness at 37 °C for 12 d |
| Mycelia with hyphal knots (Knot) | Continuous darkness at 37 °C for 5.5 d, and 12 h:12 h light–dark cycle for 1 d |
| Primordia undergoing meiosis (Pri) | Continuous darkness at 37 °C for 5.5 d, and 12 h:12 h light–dark cycle for 6 d |
| Young fruiting bodies undergoing spore formation (YFBs) | Continuous darkness at 37 °C for 5.5 d, and 12 h:12 h light–dark cycle for 6.5 d |
| Mature basidiospores (BS) | Basidiospore discharged from mature cap |
| Half-germinating basidiospores (BS12h) | Continuous darkness at 37 °C for 12 h, broth 150 rpm |
| Fully germinated basidiospores (BS24h) | Continuous darkness at 37 °C for 24 h, broth 150 rpm |
| Genomic DNA | Continuous darkness at 37 °C |
| Stage | Alternative Splicing Sites | RNA Editing Sites |
|---|---|---|
| BS | 3239 | 24 |
| BS12h | 1972 | 7 |
| BS24h | 1892 | 17 |
| Myc | 2575 | 29 |
| Oidia | 2412 | 24 |
| Scl | 2619 | 141 |
| Knot | 2829 | 33 |
| Pri | 2710 | 21 |
| YFB | 2865 | 22 |
| Total | 6839 | 217 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Y.; Chan, P.-L.; Kwan, H.-S.; Chang, J. The Genome-Wide Characterization of Alternative Splicing and RNA Editing in the Development of Coprinopsis cinerea. J. Fungi 2023, 9, 915. https://doi.org/10.3390/jof9090915
Xie Y, Chan P-L, Kwan H-S, Chang J. The Genome-Wide Characterization of Alternative Splicing and RNA Editing in the Development of Coprinopsis cinerea. Journal of Fungi. 2023; 9(9):915. https://doi.org/10.3390/jof9090915
Chicago/Turabian StyleXie, Yichun, Po-Lam Chan, Hoi-Shan Kwan, and Jinhui Chang. 2023. "The Genome-Wide Characterization of Alternative Splicing and RNA Editing in the Development of Coprinopsis cinerea" Journal of Fungi 9, no. 9: 915. https://doi.org/10.3390/jof9090915