
Global Gene Expression of Post-Senescent Telomerase-Negative ter1Δ Strain of Ustilago maydis
 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Culture Medium
2.2. RNA Isolation and Transcriptome Sequencing
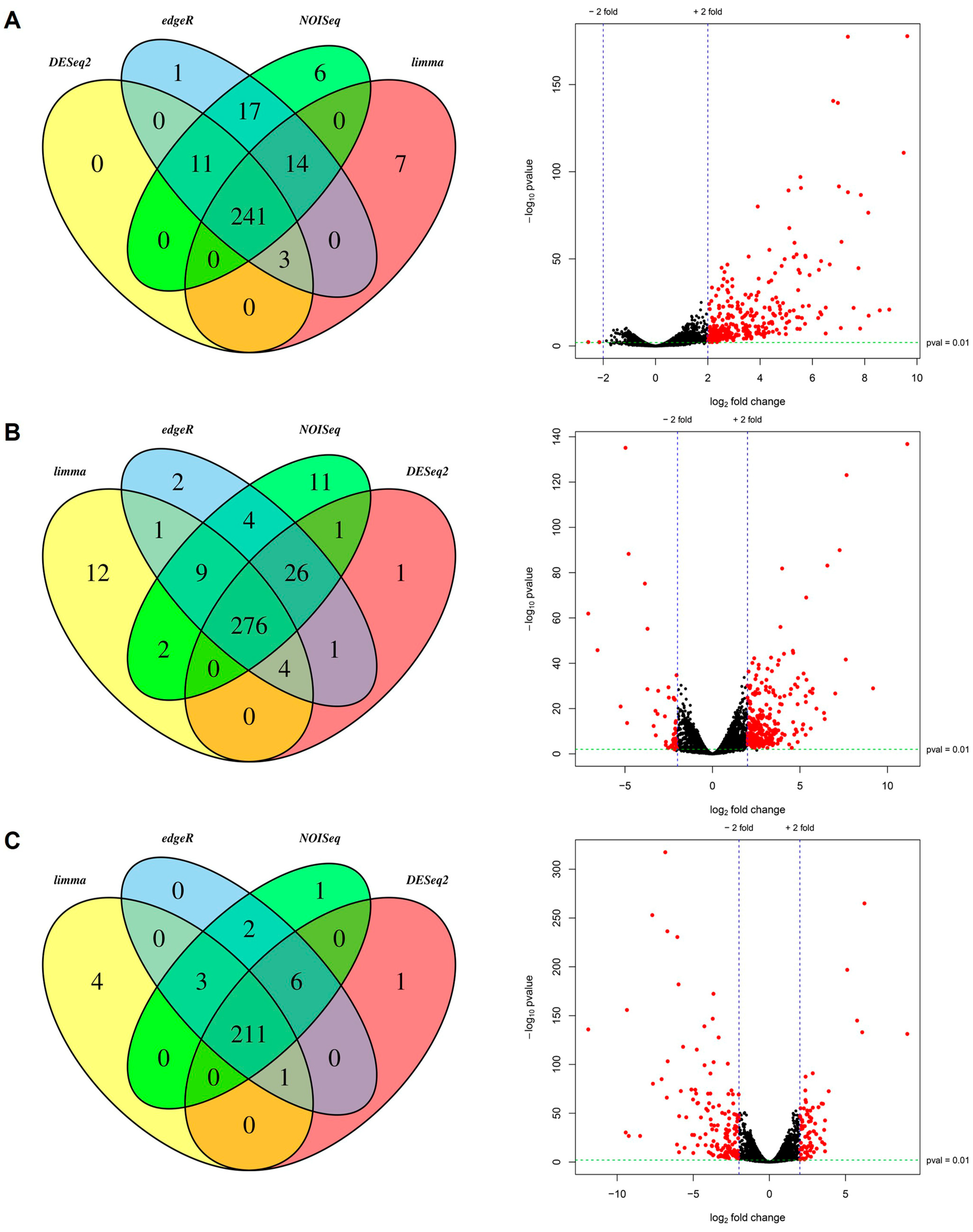
2.3. Data Analysis
2.4. Annotation and Classification of Differentially Expressed Transcripts
3. Results
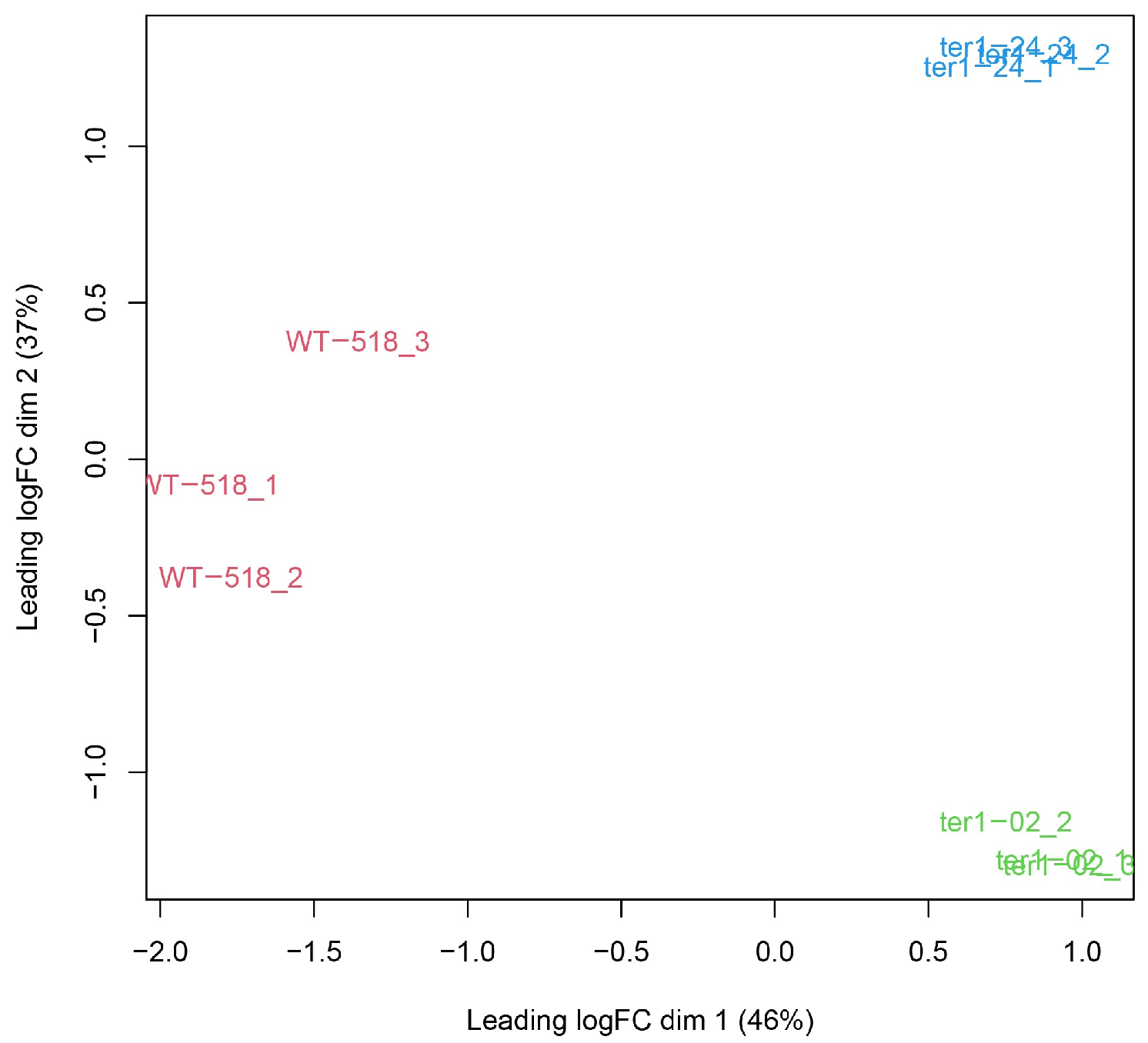
3.1. Sequencing and Quality Control of RNA-Seq Libraries
3.2. Changes in the Expression Profile of Telomerase-RNA-Deletion Response (TDR) Genes
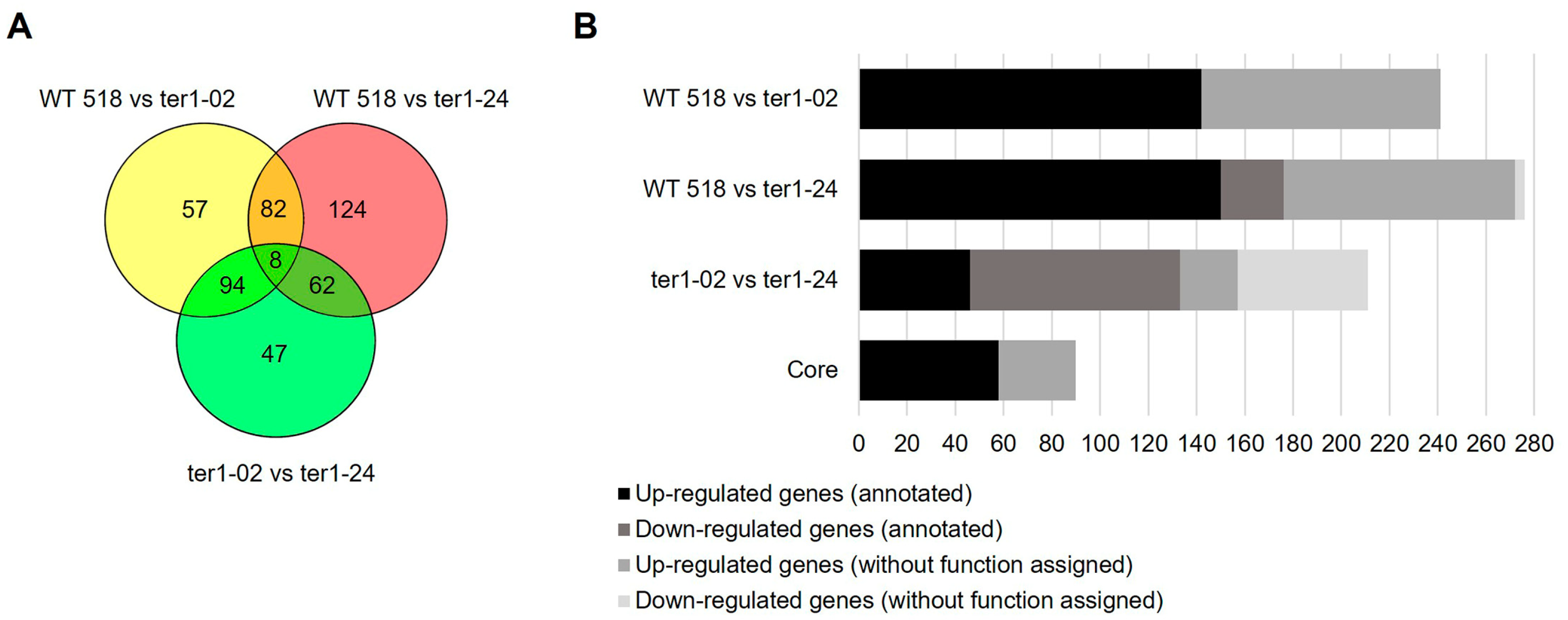
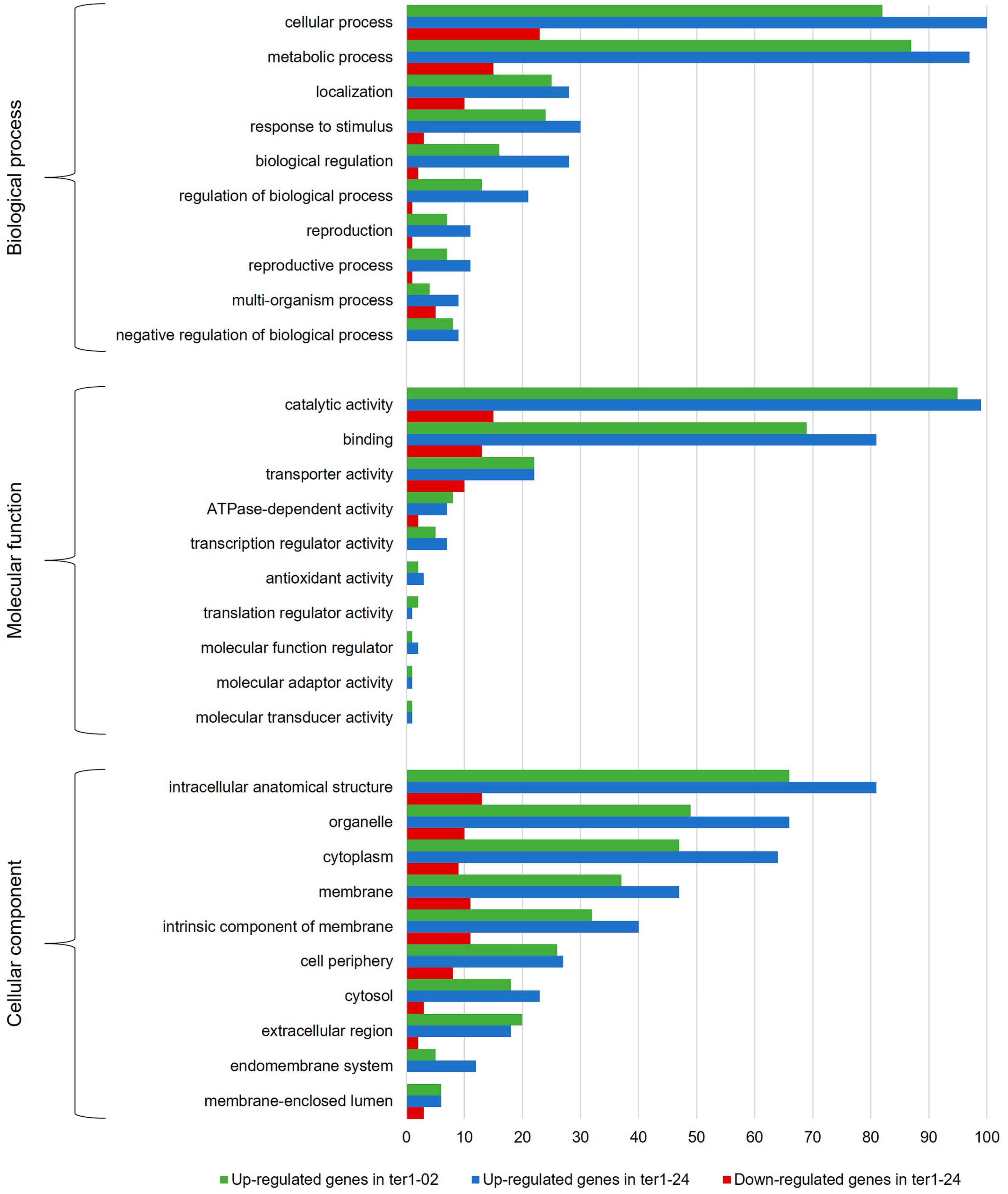
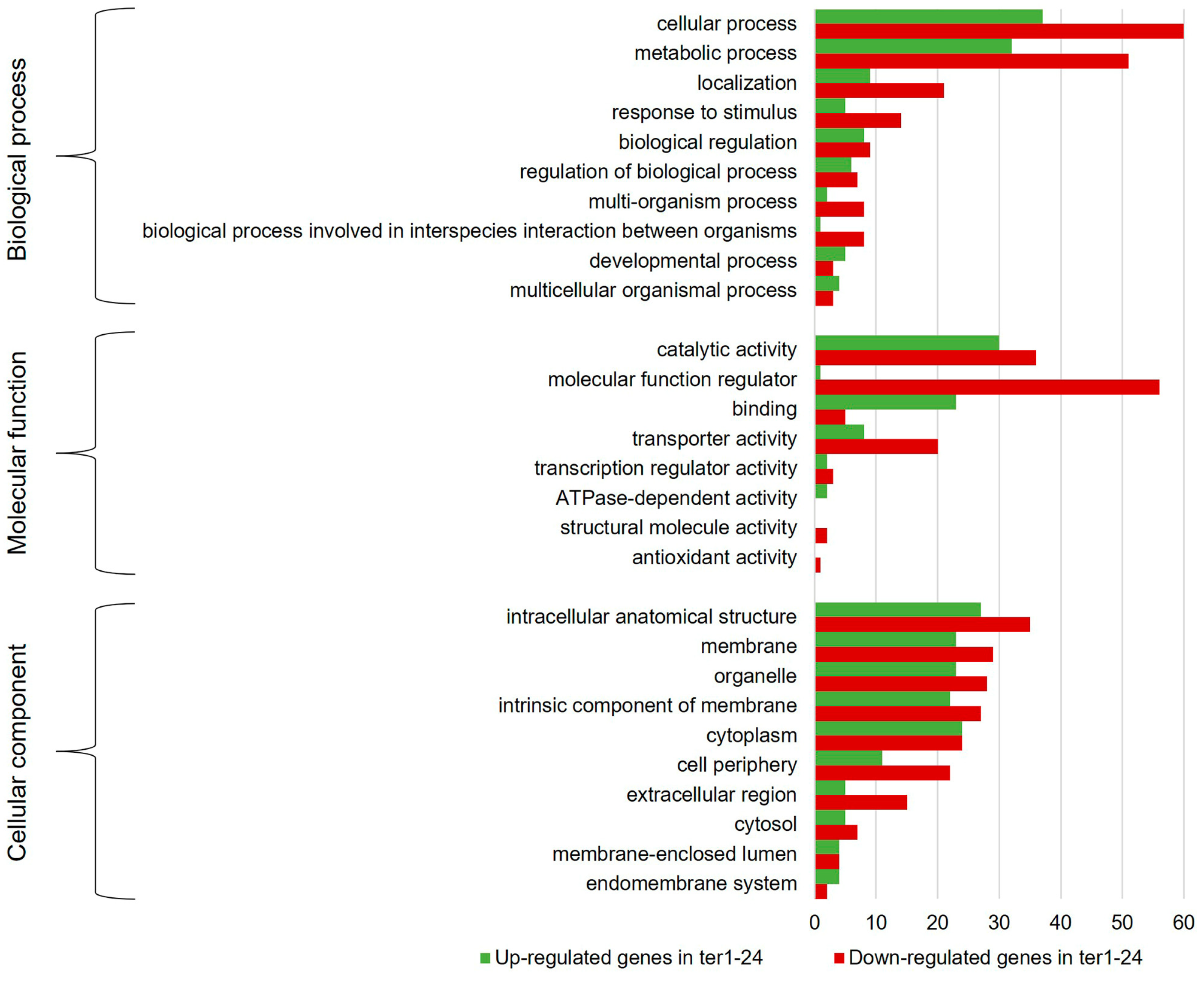
3.3. Annotation and Functional Assignment
3.4. Chromatin Structure
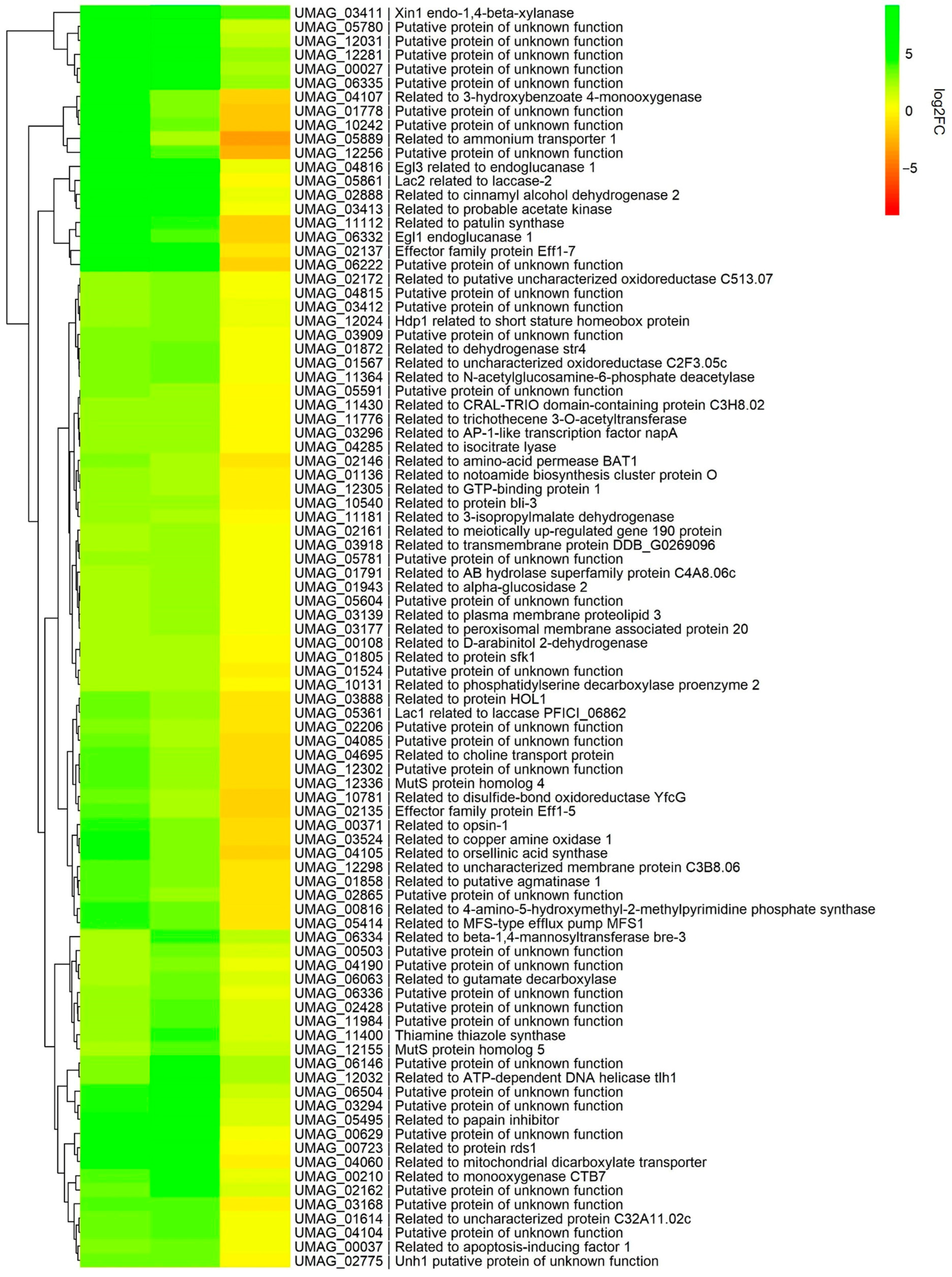
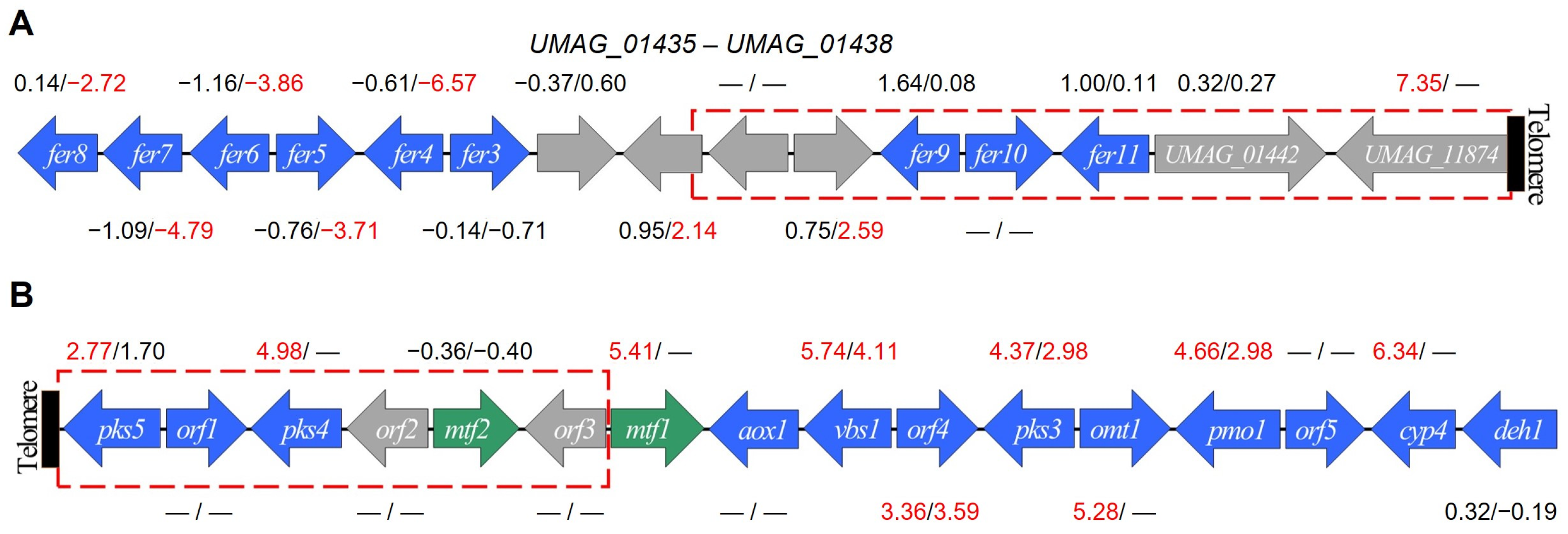
3.5. Subtelomeric Genes
3.6. DEGs Related to Stress and DNA-Damage Response (DDR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Id | Description | WT 518 vs. ter1-02 | WT 518 vs. ter1-24 | ter1-02 vs. ter1-24 |
|---|---|---|---|---|
| UMAG_12155 | Msh5—MutS protein homolog 5 2 | 2.426 | 3.797 | 1.398 |
| UMAG_12336 | Msh4—MutS protein homolog 4 2 | 3.674 | 2.454 | −1.205 |
| UMAG_10845 | Related to G/U mismatch-specific DNA glycosylase | 1.183 | 2.608 | 1.444 |
| UMAG_05917 | Related to cryptochrome DASH | 1.200 | 2.370 | 1.199 |
| UMAG_01262 | Related to DNA polymerase beta | 1.834 | 2.397 | 0.585 |
| UMAG_03290 | Rad51—DNA repair protein RAD51 1 | 2.046 | 0.765 | −1.256 |
| UMAG_11008 | Mer3—ATP-dependent DNA helicase MER3 2 | 2.011 | 2.427 | 0.432 |
| UMAG_04165 | Related to replication factor A protein 3 | 1.741 | −0.387 | −2.098 |
| UMAG_01952 | Related to UV-damage endonuclease | 1.812 | 2.683 | 0.889 |
| UMAG_00172 | Related to meiotic recombination protein rec8 | 1.749 | 2.187 | 0.481 |
3.7. Genes Involved in Telomere Maintenance
3.7.1. Complexes of Shelterin, CST, and MRX/MRN
3.7.2. Putative SM7-like Subunits
3.7.3. Telomere-Linked Helicases (TLH1-like)
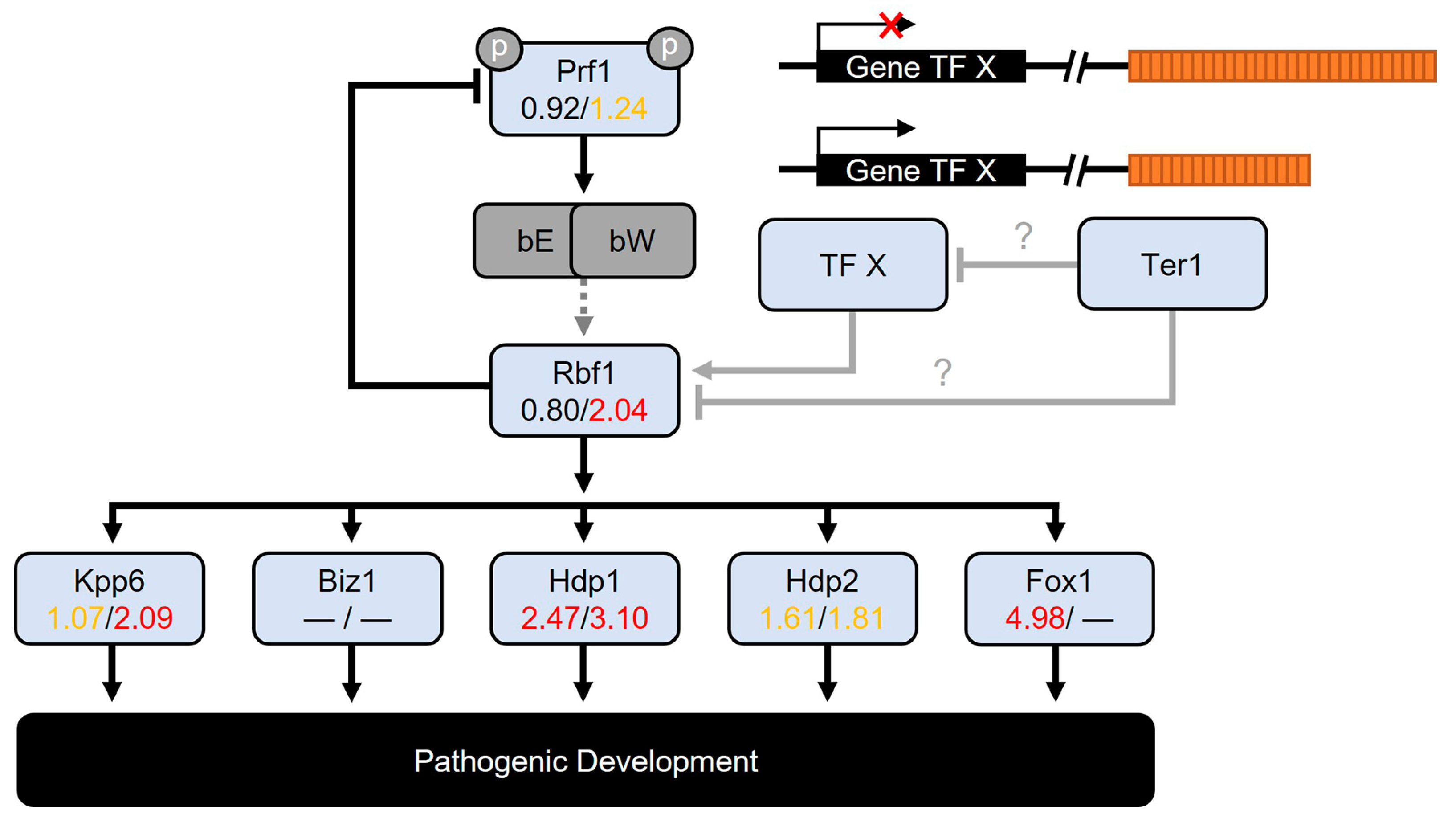
3.8. Genes Involved in Cell Cycle Progression and Pathogenic Development
3.8.1. The Cell Cycle Progression
| Gene Id | Description | WT 518 vs. ter1-02 | WT 518 vs. ter1-24 | ter1-02 vs. ter1-24 |
|---|---|---|---|---|
| UMAG_10529 | Pcl12—related to PHO85 cyclin-2 10 | 0.860 | 3.388 | 2.535 |
| UMAG_00628 | Putative protein of unknown function 8 | 5.109 | — | −1.462 |
| UMAG_00876 | Related to glucan 1,3-beta-glucosidase 1 | 0.900 | −1.435 | −2.31 |
| UMAG_01130 | Related to tyrosinase ustQ 7 | −0.164 | 2.628 | 2.801 |
| UMAG_01237 | Putative protein of unknown function, cluster 2A 1 | 5.519 | — | −5.015 |
| UMAG_01238 | Putative protein of unknown function, cluster 2A 1 | 6.249 | — | −5.815 |
| UMAG_01431 | Fer6—multidrug resistance protein fer6 2 | −1.163 | −3.861 | −2.674 |
| UMAG_01432 | Fer5—acyltransferase fer5 2 | −0.767 | −3.711 | −2.92 |
| UMAG_01433 | Fer4—enoyl-CoA isomerase/hydratase fer4 2 | −0.613 | −6.571 | −5.924 |
| UMAG_01690 | Putative protein of unknown function 7 | 3.949 | — | −2.207 |
| UMAG_01695 | Stp6—Putative protein of unknown function 10 | — | 5.615 | 0.842 |
| UMAG_01788 | Related to chitin deacetylase 3 | 1.022 | −2.465 | −3.476 |
| UMAG_01829 | Related to alpha-L-arabinofuranosidase A 1 | 2.673 | 1.529 | −1.126 |
| UMAG_01888 | Probable serine carboxypeptidase, cluster 3A 1 | 0.532 | 2.143 | 1.633 |
| UMAG_01945 | Putative invertase 3 | 2.373 | 4.817 | 2.465 |
| UMAG_02135 | Effector family protein Eff1-5 6 | 3.740 | 2.215 | −1.498 |
| UMAG_02136 | Effector family protein Eff1-6 6 | 7.088 | — | −1.341 |
| UMAG_02137 | Effector family protein Eff1-7 6 | 5.394 | 4.716 | −0.649 |
| UMAG_02138 | Effector family protein Eff1-8 6 | 3.099 | 2.239 | −0.820 |
| UMAG_02140 | Effector family protein Eff1-10 6 | 2.426 | 2.692 | 0.301 |
| UMAG_02758 | Putative protein of unknown function 3 | −0.473 | −2.133 | −1.632 |
| UMAG_03023 | Related to ribonuclease T2-like 1-A 3 | −0.653 | −4.881 | −4.193 |
| UMAG_03382 * | Related to 3-phytase A 3 | 1.305 | 2.814 | 1.534 |
| UMAG_03411 | Xin1—endo-1,4-beta-xylanase 1 | 5.562 | 9.175 | 3.65 |
| UMAG_03416 | Putative protein of unknown function 1 | 2.482 | 2.625 | 0.160 |
| UMAG_03749 | Putative protein of unknown function, cluster 10A 1 | 1.944 | 2.601 | 0.700 |
| UMAG_03750 | Putative protein of unknown function, cluster 10A 1 | — | 4.125 | 2.386 |
| UMAG_03751 | Putative protein of unknown function, cluster 10A 1 | 5.598 | 3.721 | −1.824 |
| UMAG_04282 | Related to 3-phytase A 3 | 1.336 | 3.869 | 2.556 |
| UMAG_04309 | Probable alpha-L-arabinofuranosidase 1 | 4.469 | 0.820 | −3.625 |
| UMAG_04364 | Related to glucan 1,3-beta-glucosidase 1 | 2.904 | 2.711 | −0.162 |
| UMAG_04503 | Probable alpha-galactosidase D 1 | 4.467 | — | −3.390 |
| UMAG_04816 | Egl3—Related to endoglucanase 1 1, 5 | 5.717 | 6.415 | 0.698 |
| UMAG_05036 | Related to probable glycosidase C21B10.07 3 | 0.744 | 3.080 | 2.360 |
| UMAG_05299 | Putative protein of unknown function, cluster 19A 1 | 0.756 | 2.789 | 2.067 |
| UMAG_05306 | Putative protein of unknown function, cluster 19A 1 | 4.330 | — | −1.603 |
| UMAG_05308 | Putative protein of unknown function, cluster 19A 1 | 2.876 | — | −2.949 |
| UMAG_05310 | Putative protein of unknown function, cluster 19A 1 | 7.574 | — | −9.239 |
| UMAG_05314 | Putative protein of unknown function, cluster 19A 1 | 3.579 | — | −3.060 |
| UMAG_05361 | Lac1—laccase 3, 4 | 3.437 | 2.576 | −0.836 |
| UMAG_05439 | Related to GlcNAc-binding protein A 7 | 3.655 | −0.031 | −3.665 |
| UMAG_05495 | Related to papain inhibitor 7 | 4.355 | 5.372 | 1.037 |
| UMAG_05689 | Related to Fe-regulated protein 8 2 | 2.425 | 1.702 | −0.700 |
| UMAG_05861 | Lac2—laccase-2 3, 4 | 5.316 | 5.355 | 0.064 |
| UMAG_06190 | Related to chitinase A1 3 | 3.479 | — | −1.840 |
| UMAG_06221 | Putative protein of unknown function, cluster 22A 1 | 2.243 | 1.030 | −1.183 |
| UMAG_06222 | Putative protein of unknown function, cluster 22A 1 | 6.296 | 4.759 | −1.521 |
| UMAG_06274 | Related to hormone-sensitive lipase 3 | 1.259 | 3.221 | 1.985 |
| UMAG_06332 | Egl1—endoglucanase 1 1, 5 | 5.283 | 3.747 | −1.518 |
| UMAG_10055 | Related to glutathione hydrolase proenzyme 3 | 3.490 | — | −2.091 |
| UMAG_10557 | Putative protein of unknown function, cluster 19A 1 | 4.454 | — | −3.768 |
| UMAG_11338 | Fer8—Fe-regulated protein 8 2 | 0.141 | −2.720 | −2.833 |
| UMAG_11339 | Fer7—siderophore transporter fer7 2 | −1.095 | −4.791 | −3.672 |
| UMAG_12330 | Putative protein of unknown function 3 | 1.550 | 3.223 | 1.701 |
| UMAG_05528 | Related to alkali-sensitive linkage protein 1 9 | 5.131 | — | −4.945 |
| UMAG_02161 | Related to meiotically upregulated gene 190 protein | 2.322 | 2.503 | 0.205 |
| UMAG_02517 | Gpa2—guanine nucleotide-binding protein alpha-2 subunit | 1.110 | 2.444 | 1.360 |
| UMAG_02994 | Related to sporulation-specific protein 5 | −0.065 | 2.505 | 2.591 |
| UMAG_03541 | Related to meiotic expression upregulated protein 26 | 4.233 | — | −0.999 |
| UMAG_05467 | Related to meiotic coiled-coil protein 2 | 5.627 | — | −3.148 |
| UMAG_11677 | Related to serine/threonine-protein kinase cek1 | 0.837 | 2.533 | 1.720 |
3.8.2. The Pathogenic Development
3.9. Differentially Expressed Transcription Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pfeiffer, V.; Lingner, J. Replication of Telomeres and the Regulation of Telomerase. Cold Spring Harb. Perspect. Biol. 2013, 5, a010405. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V. The End Replication Problem: More Than One Solution. Nat. Med. 1997, 3, 1198–1199. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Collins, K. Telomerase: An Rnp Enzyme Synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, a003558. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Benetti, R.; Blasco, M.A. Telomerase Regulation and Stem Cell Behaviour. Curr. Opin. Cell Biol. 2006, 18, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Flores, I.; Canela, A.; Vera, E.; Tejera, A.; Cotsarelis, G.; Blasco, M.A. The Longest Telomeres: A General Signature of Adult Stem Cell Compartments. Genes Dev. 2008, 22, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Taggart, A.K.; Teng, S.C.; Zakian, V.A. Est1p as a Cell Cycle-Regulated Activator of Telomere-Bound Telomerase. Science 2002, 297, 1023–1026. [Google Scholar] [CrossRef]
- Mozdy, A.D.; Cech, T.R. Low Abundance of Telomerase in Yeast: Implications for Telomerase Haploinsufficiency. Rna 2006, 12, 1721–1737. [Google Scholar] [CrossRef]
- Dionne, I.; Larose, S.; Dandjinou, A.T.; Abou Elela, S.; Wellinger, R.J. Cell Cycle-Dependent Transcription Factors Control the Expression of Yeast Telomerase RNA. RNA 2013, 19, 992–1002. [Google Scholar] [CrossRef]
- Singer, M.S.; Gottschling, D.E. Tlc1: Template RNA Component of Saccharomyces Cerevisiae Telomerase. Science 1994, 266, 404–409. [Google Scholar] [CrossRef]
- IJpma, A.S.; Greider, C.W. Short Telomeres Induce a DNA Damage Response in Saccharomyces Cerevisiae. Mol Biol Cell 2003, 14, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, V.; Blackburn, E.H. An Alternative Pathway for Yeast Telomere Maintenance Rescues Est7- Senescence. Cell 1993, 73, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.C.; Zakian, V.A. Telomere-Telomere Recombination Is an Efficient Bypass Pathway for Telomere Maintenance in Saccharomyces Cerevisiae. Mol. Cell Biol. 1999, 19, 8083–8093. [Google Scholar] [CrossRef] [PubMed]
- Makovets, S.; Williams, T.L.; Blackburn, E.H. The Telotype Defines the Telomere State in Saccharomyces Cerevisiae and Is Inherited as a Dominant Non-Mendelian Characteristic in Cells Lacking Telomerase. Genetics 2008, 178, 245–257. [Google Scholar] [CrossRef]
- Hu, Y.; Tang, H.B.; Liu, N.N.; Tong, X.J.; Dang, W.; Duan, Y.M.; Fu, X.H.; Zhang, Y.; Peng, J.; Meng, F.L.; et al. Telomerase-Null Survivor Screening Identifies Novel Telomere Recombination Regulators. PLoS Genet. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Nautiyal, S.; DeRisi, J.L.; Blackburn, E.H. The Genome-Wide Expression Response to Telomerase Deletion in Saccharomyces Cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 9316–9321. [Google Scholar] [CrossRef]
- Chen, D.; Toone, W.M.; Mata, J.; Lyne, R.; Burns, G.; Kivinen, K.; Brazma, A.; Jones, N.; Bähler, J. Global Transcriptional Responses of Fission Yeast to Environmental Stress. Mol. Biol. Cell 2003, 14, 214–229. [Google Scholar] [CrossRef]
- Beletsky, A.V.; Malyavko, A.N.; Sukhanova, M.V.; Mardanova, E.S.; Zvereva, M.I.; Petrova, O.A.; Parfenova, Y.Y.; Rubtsova, M.P.; Mardanov, A.V.; Lavrik, O.I.; et al. The Genome-Wide Transcription Response to Telomerase Deficiency in the Thermotolerant Yeast Hansenula Polymorpha Dl-1. BMC Genom. 2017, 18, 492. [Google Scholar] [CrossRef]
- Červenák, F.; Juríková, K.; Devillers, H.; Kaffe, B.; Khatib, A.; Bonnell, E.; Sopkovičová, M.; Wellinger, R.J.; Nosek, J.; Tzfati, Y.; et al. Identification of Telomerase RNAs in Species of the Yarrowia Clade Provides Insights into the Co-Evolution of Telomerase, Telomeric Repeats and Telomere-Binding Proteins. Sci. Rep. 2019, 9, 13365. [Google Scholar] [CrossRef]
- Niederer, R.O.; Papadopoulos, N.; Zappulla, D.C. Identification of Novel Noncoding Transcripts in Telomerase-Negative Yeast Using RNA-Seq. Sci. Rep. 2016, 6, 19376. [Google Scholar] [CrossRef]
- Guzman, P.A.; Sanchez, J.G. Characterization of Telomeric Regions from Ustilago maydis. Microbiology 1994, 140 Pt 3, 551–557. [Google Scholar] [CrossRef]
- Bautista-Espana, D.; Anastacio-Marcelino, E.; Horta-Valerdi, G.; Celestino-Montes, A.; Kojic, M.; Negrete-Abascal, E.; Reyes-Cervantes, H.; Vazquez-Cruz, C.; Guzman, P.; Sanchez-Alonso, P. The Telomerase Reverse Transcriptase Subunit from the Dimorphic Fungus Ustilago maydis. PLoS ONE 2014, 9, e109981. [Google Scholar] [CrossRef]
- Yu, E.Y.; Kojic, M.; Holloman, W.K.; Lue, N.F. Brh2 and Rad51 Promote Telomere Maintenance in Ustilago maydis, a New Model System of DNA Repair Proteins at Telomeres. DNA Repair. 2013, 12, 472–479. [Google Scholar] [CrossRef]
- Logeswaran, D.; Li, Y.; Akhter, K.; Podlevsky, J.D.; Olson, T.L.; Forsberg, K.; Chen, J.J. Biogenesis of Telomerase RNA from a Protein-Coding Mrna Precursor. Proc. Natl. Acad. Sci. USA 2022, 119, e2204636119. [Google Scholar] [CrossRef] [PubMed]
- Sanpedro-Luna, J.A.; Jacinto-Vázquez, J.J.; Anastacio-Marcelino, E.; Posadas-Gutiérrez, C.M.; Olmos-Pineda, I.; González-Bernal, J.A.; Carcaño-Montiel, M.; Vega-Alvarado, L.; Vázquez-Cruz, C.; Sánchez-Alonso, P. Telomerase RNA Plays a Major Role in the Completion of the Life Cycle in Ustilago maydis and Shares Conserved Domains with Other Ustilaginales. PLoS ONE 2023, 18, e0281251. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, P.; Guzman, P. Predicted Elements of Telomere Organization and Function in Ustilago maydis. Fungal. Genet. Biol. 2008, 45 (Suppl. S1), S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. Rsem: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Jiménez-Jacinto, V.; Sanchez-Flores, A.; Vega-Alvarado, L. Integrative Differential Expression Analysis for Multiple Experiments (Ideamex): A Web Server Tool for Integrated RNA-Seq Data Analysis. Front. Genet. 2019, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for Rna-Seq Data with Deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential Expression in RNA-Seq: A Matter of Depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. Hmmer Web Server: Interactive Sequence Similarity Searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Donaldson, M.E.; Ostrowski, L.A.; Goulet, K.M.; Saville, B.J. Transcriptome Analysis of Smut Fungi Reveals Widespread Intergenic Transcription and Conserved Antisense Transcript Expression. BMC Genom. 2017, 18, 340. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alonso, P.; Guzman, P. Organization of Chromosome Ends in Ustilago maydis. Recq-Like Helicase Motifs at Telomeric Regions. Genetics 1998, 148, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Dutheil, J.Y.; Münch, K.; Schotanus, K.; Stukenbrock, E.H.; Kahmann, R. The Insertion of a Mitochondrial Selfish Element into the Nuclear Genome and Its Consequences. Ecol. Evol. 2020, 10, 11117–11132. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Khang, C.H.; Park, S.Y.; Lee, Y.H.; Kang, S. Evolution and Organization of a Highly Dynamic, Subtelomeric Helicase Gene Family in the Rice Blast Fungus Magnaporthe Grisea. Genetics 2002, 162, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.R.; Ibarra, P.T.; Thon, G. Evolutionary-Conserved Telomere-Linked Helicase Genes of Fission Yeast Are Repressed by Silencing Factors, Rnai Components and the Telomere-Binding Protein Taz1. Nucleic Acids Res. 2006, 34, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Kamper, J.; Kahmann, R.; Bolker, M.; Ma, L.J.; Brefort, T.; Saville, B.J.; Banuett, F.; Kronstad, J.W.; Gold, S.E.; Muller, O.; et al. Insights from the Genome of the Biotrophic Fungal Plant Pathogen Ustilago maydis. Nature 2006, 444, 97–101. [Google Scholar] [CrossRef]
- Ross-Macdonald, P.; Roeder, G.S. Mutation of a Meiosis-Specific Muts Homolog Decreases Crossing over but Not Mismatch Correction. Cell 1994, 79, 1069–1080. [Google Scholar] [CrossRef]
- Hollingsworth, N.M.; Ponte, L.; Halsey, C. Msh5, a Novel Muts Homolog, Facilitates Meiotic Reciprocal Recombination between Homologs in Saccharomyces Cerevisiae but Not Mismatch Repair. Genes Dev. 1995, 9, 1728–1739. [Google Scholar] [CrossRef]
- Lynn, A.; Soucek, R.; Börner, G.V. Zmm Proteins During Meiosis: Crossover Artists at Work. Chromosome Res. 2007, 15, 591–605. [Google Scholar] [CrossRef]
- Ferguson, D.O.; Rice, M.C.; Rendi, M.H.; Kotani, H.; Kmiec, E.B.; Holloman, W.K. Interaction between Ustilago maydis Rec2 and Rad51 Genes in DNA Repair and Mitotic Recombination. Genetics 1997, 145, 243–251. [Google Scholar] [CrossRef]
- Holloman, W.K.; Schirawski, J.; Holliday, R. The Homologous Recombination System of Ustilago maydis. Fungal Genet. Biol. 2008, 45 (Suppl. S1), S31–S39. [Google Scholar] [CrossRef]
- Mandell, J.G.; Goodrich, K.J.; Bähler, J.; Cech, T.R. Expression of a Recq Helicase Homolog Affects Progression through Crisis in Fission Yeast Lacking Telomerase. J. Biol. Chem. 2005, 280, 5249–5257. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martin, J.; Castillo-Lluva, S.; Sgarlata, C.; Flor-Parra, I.; Mielnichuk, N.; Torreblanca, J.; Carbo, N. Pathocycles: Ustilago maydis as a Model to Study the Relationships between Cell Cycle and Virulence in Pathogenic Fungi. Mol. Genet. Genom. MGG 2006, 276, 211–229. [Google Scholar] [CrossRef]
- Banuett, F.; Herskowitz, I. Discrete Developmental Stages During Teliospore Formation in the Corn Smut Fungus, Ustilago maydis. Development 1996, 122, 2965–2976. [Google Scholar] [CrossRef] [PubMed]
- Skibbe, D.S.; Doehlemann, G.; Fernandes, J.; Walbot, V. Maize Tumors Caused by Ustilago maydis Require Organ-Specific Genes in Host and Pathogen. Science 2010, 328, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Doehlemann, G.; van der Linde, K.; Aßmann, D.; Schwammbach, D.; Hof, A.; Mohanty, A.; Jackson, D.; Kahmann, R. Pep1, a Secreted Effector Protein of Ustilago maydis, Is Required for Successful Invasion of Plant Cells. PLoS Pathog. 2009, 5, e1000290. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Steinberg, G.; Pérez-Martín, J. Characterization of B-Type Cyclins in the Smut Fungus Ustilago maydis: Roles in Morphogenesis and Pathogenicity. J. Cell Sci. 2004, 117, 487–506. [Google Scholar] [CrossRef]
- Castillo-Lluva, S.; Alvarez-Tabarés, I.; Weber, I.; Steinberg, G.; Pérez-Martín, J. Sustained Cell Polarity and Virulence in the Phytopathogenic Fungus Ustilago maydis Depends on an Essential Cyclin-Dependent Kinase from the Cdk5/Pho85 Family. J. Cell Sci. 2007, 120, 1584–1595. [Google Scholar] [CrossRef]
- Flor-Parra, I.; Castillo-Lluva, S.; Perez-Martin, J. Polar Growth in the Infectious Hyphae of the Phytopathogen Ustilago maydis Depends on a Virulence-Specific Cyclin. Plant Cell 2007, 19, 3280–3296. [Google Scholar] [CrossRef]
- Martinez-Soto, D.; Robledo-Briones, A.M.; Estrada-Luna, A.A.; Ruiz-Herrera, J. Transcriptomic Analysis of Ustilago maydis Infecting Arabidopsis Reveals Important Aspects of the Fungus Pathogenic Mechanisms. Plant Signal. Behav. 2013, 8, e25059. [Google Scholar] [CrossRef]
- Eichhorn, H.; Lessing, F.; Winterberg, B.; Schirawski, J.; Kamper, J.; Muller, P.; Kahmann, R. A Ferroxidation/Permeation Iron Uptake System Is Required for Virulence in Ustilago maydis. Plant Cell 2006, 18, 3332–3345. [Google Scholar] [CrossRef]
- Mueller, O.; Kahmann, R.; Aguilar, G.; Trejo-Aguilar, B.; Wu, A.; de Vries, R.P. The Secretome of the Maize Pathogen Ustilago maydis. Fungal Genet. Biol. 2008, 45 (Suppl. S1), S63–S70. [Google Scholar] [CrossRef]
- Doyle, C.E.; Kitty Cheung, H.Y.; Spence, K.L.; Saville, B.J. Unh1, an Ustilago maydis Ndt80-Like Protein, Controls Completion of Tumor Maturation, Teliospore Development, and Meiosis. Fungal Genet. Biol. 2016, 94, 54–68. [Google Scholar] [CrossRef]
- Lanver, D.; Berndt, P.; Tollot, M.; Naik, V.; Vranes, M.; Warmann, T.; Munch, K.; Rossel, N.; Kahmann, R. Plant Surface Cues Prime Ustilago maydis for Biotrophic Development. PLoS Pathog. 2014, 10, e1004272. [Google Scholar] [CrossRef]
- Khrunyk, Y.; Munch, K.; Schipper, K.; Lupas, A.N.; Kahmann, R. The Use of Flp-Mediated Recombination for the Functional Analysis of an Effector Gene Family in the Biotrophic Smut Fungus Ustilago maydis. New Phytol. 2010, 187, 957–968. [Google Scholar] [CrossRef]
- Schilling, L.; Matei, A.; Redkar, A.; Walbot, V.; Doehlemann, G. Virulence of the Maize Smut Ustilago maydis Is Shaped by Organ-Specific Effectors. Mol. Plant Pathol. 2014, 15, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Matei, A.; Ernst, C.; Günl, M.; Thiele, B.; Altmüller, J.; Walbot, V.; Usadel, B.; Doehlemann, G. How to Make a Tumour: Cell Type Specific Dissection of Ustilago maydis-Induced Tumour Development in Maize Leaves. New Phytol. 2018, 217, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Okmen, B.; Doehlemann, G. Inside Plant: Biotrophic Strategies to Modulate Host Immunity and Metabolism. Curr. Opin. Plant Biol. 2014, 20, 19–25. [Google Scholar] [CrossRef]
- Kojic, M.; Kostrub, C.F.; Buchman, A.R.; Holloman, W.K. Brca2 Homolog Required for Proficiency in DNA Repair, Recombination, and Genome Stability in Ustilago maydis. Mol. Cell 2002, 10, 683–691. [Google Scholar] [CrossRef]
- Mielnichuk, N.; Sgarlata, C.; Perez-Martin, J. A Role for the DNA-Damage Checkpoint Kinase Chk1 in the Virulence Program of the Fungus Ustilago maydis. J. Cell Sci. 2009, 122, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Tomonaga, T.; Nagao, K.; Kawasaki, Y.; Furuya, K.; Murakami, A.; Morishita, J.; Yuasa, T.; Sutani, T.; Kearsey, S.E.; Uhlmann, F.; et al. Characterization of Fission Yeast Cohesin: Essential Anaphase Proteolysis of Rad21 Phosphorylated in the S Phase. Genes Dev. 2000, 14, 2757–2770. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Tomaszewski, J.M.; Vazquez, M.; Verschoor, S.; et al. Rad21-Cohesin Haploinsufficiency Impedes DNA Repair and Enhances Gastrointestinal Radiosensitivity in Mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. Rad21 Mutations Cause a Human Cohesinopathy. Am. J. Hum. Genet 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Reissmann, S.; Schipper, K.; Gonzalez, C.; Assmann, D.; Glatter, T.; Moretti, M.; Ma, L.S.; Rexer, K.H.; Snetselaar, K.; et al. A Cell Surface-Exposed Protein Complex with an Essential Virulence Function in Ustilago maydis. Nat. Microbiol. 2021, 6, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Fernández, E.Z.; Shi, Y.M.; Grün, P.; Bode, H.B.; Bölker, M. An Unconventional Melanin Biosynthesis Pathway in Ustilago maydis. Appl. Environ. Microbiol. 2021, 87, e01510-20. [Google Scholar] [CrossRef]
- Mei, B.; Budde, A.D.; Leong, S.A. Sid1, a Gene Initiating Siderophore Biosynthesis in Ustilago maydis: Molecular Characterization, Regulation by Iron, and Role in Phytopathogenicity. Proc. Natl. Acad Sci. USA 1993, 90, 903–907. [Google Scholar] [CrossRef]
- Pothiratana, C. Functional Characterization of the Homeodomain Transcription Factor Hdp1 in Ustilago maydis. Ph.D. Thesis, Philipps-Universität Marburg, Marburg, Germany, 2007; p. 105. [Google Scholar]
- Zahiri, A.R.; Babu, M.R.; Saville, B.J. Differential Gene Expression During Teliospore Germination in Ustilago maydis. Mol. Genet. Genom. 2005, 273, 394–403. [Google Scholar] [CrossRef]
- Heimel, K.; Scherer, M.; Vranes, M.; Wahl, R.; Pothiratana, C.; Schuler, D.; Vincon, V.; Finkernagel, F.; Flor-Parra, I.; Kamper, J. The Transcription Factor Rbf1 Is the Master Regulator for B-Mating Type Controlled Pathogenic Development in Ustilago maydis. PLoS Pathog. 2010, 6, e1001035. [Google Scholar] [CrossRef]
- Lanver, D.; Muller, A.N.; Happel, P.; Schweizer, G.; Haas, F.B.; Franitza, M.; Pellegrin, C.; Reissmann, S.; Altmuller, J.; Rensing, S.A.; et al. The Biotrophic Development of Ustilago maydis Studied by RNA-Seq Analysis. Plant Cell 2018, 30, 300–323. [Google Scholar] [CrossRef]
- Pinter, N.; Hach, C.A.; Hampel, M.; Rekhter, D.; Zienkiewicz, K.; Feussner, I.; Poehlein, A.; Daniel, R.; Finkernagel, F.; Heimel, K. Signal Peptide Peptidase Activity Connects the Unfolded Protein Response to Plant Defense Suppression by Ustilago maydis. PLoS Pathog. 2019, 15, e1007734. [Google Scholar] [CrossRef]
- Asano, Y.; Hagiwara, D.; Yamashino, T.; Mizuno, T. Characterization of the Bzip-Type Transcription Factor Napa with Reference to Oxidative Stress Response in Aspergillus Nidulans. Biosci. Biotechnol. Biochem. 2007, 71, 1800–1803. [Google Scholar] [CrossRef]
- Lev, S.; Hadar, R.; Amedeo, P.; Baker, S.E.; Yoder, O.C.; Horwitz, B.A. Activation of an Ap1-Like Transcription Factor of the Maize Pathogen Cochliobolus Heterostrophus in Response to Oxidative Stress and Plant Signals. Eukaryot. Cell 2005, 4, 443–454. [Google Scholar] [CrossRef] [PubMed]
- León-Ramírez, C.G.; Sánchez-Arreguin, J.A.; Cabrera-Ponce, J.L.; Martínez-Soto, D.; Ortiz-Castellanos, M.L.; Aréchiga-Carvajal, E.T.; Salazar-Chávez, M.F.; Sánchez-Segura, L.; Ruiz-Herrera, J. Tec1, a Member of the Tea Transcription Factors Family, Is Involved in Virulence and Basidiocarp Development in Ustilago maydis. Int. Microbiol. 2022, 25, 17–26. [Google Scholar] [CrossRef]
- Donaldson, M.E.; Meng, S.; Gagarinova, A.; Babu, M.; Lambie, S.C.; Swiadek, A.A.; Saville, B.J. Investigating the Ustilago maydis/Zea Mays Pathosystem: Transcriptional Responses and Novel Functional Aspects of a Fungal Calcineurin Regulatory B Subunit. Fungal Genet. Biol. 2013, 58–59, 91–104. [Google Scholar] [CrossRef]
- Lee, W. Comprehensive Discovery of Fungal Gene Clusters: Unexpected Co-Work Reflected at the Genomic Level. Ph.D. Thesis, Fakultät Wissenschaftszentrum Weihenstephan, Freising, Germany, 2020; p. 125. [Google Scholar]
- Islamovic, E.; García-Pedrajas, M.D.; Chacko, N.; Andrews, D.L.; Covert, S.F.; Gold, S.E. Transcriptome Analysis of a Ustilago maydis Ust1 Deletion Mutant Uncovers Involvement of Laccase and Polyketide Synthase Genes in Spore Development. Mol. Plant-Microbe Interact. 2015, 28, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.Y.; Perez-Martin, J.; Holloman, W.K.; Lue, N.F. Mre11 and Blm-Dependent Formation of Alt-Like Telomeres in Ku-Deficient Ustilago maydis. PLoS Genet. 2015, 11, e1005570. [Google Scholar] [CrossRef] [PubMed]
- Teng, S.C.; Epstein, C.; Tsai, Y.L.; Cheng, H.W.; Chen, H.L.; Lin, J.J. Induction of Global Stress Response in Saccharomyces Cerevisiae Cells Lacking Telomerase. Biochem. Biophys. Res. Commun. 2002, 291, 714–721. [Google Scholar] [CrossRef]
- Gottschling, D.E.; Aparicio, O.M.; Billington, B.L.; Zakian, V.A. Position Effect at S. Cerevisiae Telomeres: Reversible Repression of Pol Ii Transcription. Cell 1990, 63, 751–762. [Google Scholar] [CrossRef]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Magdinier, F.; Stadler, G.; Wagner, K.R.; Shay, J.W.; Wright, W.E. Telomere Position Effect: Regulation of Gene Expression with Progressive Telomere Shortening over Long Distances. Genes Dev. 2014, 28, 2464–2476. [Google Scholar] [CrossRef]
- Ottaviani, A.; Gilson, E.; Magdinier, F. Telomeric Position Effect: From the Yeast Paradigm to Human Pathologies? Biochimie 2008, 90, 93–107. [Google Scholar] [CrossRef]
- Azzalin, C.M.; Lingner, J. Telomere Functions Grounding on Terra Firma. Trends Cell Biol. 2015, 25, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Azzalin, C.M. The Telomeric Transcriptome: From Fission Yeast to Mammals. Int. J. Biochem. Cell Biol. 2012, 44, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Balk, B.; Maicher, A.; Dees, M.; Klermund, J.; Luke-Glaser, S.; Bender, K.; Luke, B. Telomeric Rna-DNA Hybrids Affect Telomere-Length Dynamics and Senescence. Nat. Struct. Mol. Biol. 2013, 20, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Vinayagamurthy, S.; Bagri, S.; Mergny, J.L.; Chowdhury, S. Telomeres Expand Sphere of Influence: Emerging Molecular Impact of Telomeres in Non-Telomeric Functions. Trends Genet. 2023, 39, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, A.; Paszel-Jaworska, A.; Totoń, E.; Lisiak, N.; Hołysz, H.; Królak, A.; Grodecka-Gazdecka, S.; Rubiś, B. The Non-Canonical Functions of Telomerase: To Turn Off or Not to Turn Off. Mol. Biol. Rep. 2019, 46, 1401–1411. [Google Scholar] [CrossRef]
- Ségal-Bendirdjian, E.; Geli, V. Non-Canonical Roles of Telomerase: Unraveling the Imbroglio. Front. Cell Dev. Biol. 2019, 7, 332. [Google Scholar] [CrossRef]
- Kedde, M.; le Sage, C.; Duursma, A.; Zlotorynski, E.; van Leeuwen, B.; Nijkamp, W.; Beijersbergen, R.; Agami, R. Telomerase-Independent Regulation of Atr by Human Telomerase RNA. J. Biol. Chem. 2006, 281, 40503–40514. [Google Scholar] [CrossRef]
- Eitan, E.; Tamar, A.; Yossi, G.; Peleg, R.; Braiman, A.; Priel, E. Expression of Functional Alternative Telomerase RNA Component Gene in Mouse Brain and in Motor Neurons Cells Protects from Oxidative Stress. Oncotarget 2016, 7, 78297–78309. [Google Scholar] [CrossRef]
- Gazzaniga, F.S.; Blackburn, E.H. An Antiapoptotic Role for Telomerase RNA in Human Immune Cells Independent of Telomere Integrity or Telomerase Enzymatic Activity. Blood 2014, 124, 3675–3684. [Google Scholar] [CrossRef]
- Rubtsova, M.; Naraykina, Y.; Vasilkova, D.; Meerson, M.; Zvereva, M.; Prassolov, V.; Lazarev, V.; Manuvera, V.; Kovalchuk, S.; Anikanov, N.; et al. Protein Encoded in Human Telomerase RNA Is Involved in Cell Protective Pathways. Nucleic Acids Res. 2018, 46, 8966–8977. [Google Scholar] [CrossRef]
- Li, S.; Crothers, J.; Haqq, C.M.; Blackburn, E.H. Cellular and Gene Expression Responses Involved in the Rapid Growth Inhibition of Human Cancer Cells by RNA Interference-Mediated Depletion of Telomerase Rna. J. Biol. Chem. 2005, 280, 23709–23717. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Y.; Ge, Y.; Liu, J.; Zhao, Y. Terc Promotes Cellular Inflammatory Response Independent of Telomerase. Nucleic Acids Res. 2019, 47, 8084–8095. [Google Scholar] [CrossRef] [PubMed]
- Alcaraz-Pérez, F.; García-Castillo, J.; García-Moreno, D.; López-Muñoz, A.; Anchelin, M.; Angosto, D.; Zon, L.I.; Mulero, V.; Cayuela, M.L. A Non-Canonical Function of Telomerase RNA in the Regulation of Developmental Myelopoiesis in Zebrafish. Nat. Commun. 2014, 5, 3228. [Google Scholar] [CrossRef] [PubMed]
- Jose, S.S.; Tidu, F.; Burilova, P.; Kepak, T.; Bendickova, K.; Fric, J. The Telomerase Complex Directly Controls Hematopoietic Stem Cell Differentiation and Senescence in an Induced Pluripotent Stem Cell Model of Telomeropathy. Front. Genet. 2018, 9, 345. [Google Scholar] [CrossRef]
- Romeis, T.; Brachmann, A.; Kahmann, R.; Kämper, J. Identification of a Target Gene for the Be-Bw Homeodomain Protein Complex in Ustilago maydis. Mol. Microbiol. 2000, 37, 54–66. [Google Scholar] [CrossRef]
- Reichmann, M.; Jamnischek, A.; Weinzierl, G.; Ladendorf, O.; Huber, S.; Kahmann, R.; Kamper, J. The Histone Deacetylase Hda1 from Ustilago maydis Is Essential for Teliospore Development. Mol. Microbiol. 2002, 46, 1169–1182. [Google Scholar] [CrossRef]
- Cabrita, A.; David-Palma, M.; Brito, P.H.; Heitman, J.; Coelho, M.A.; Gonçalves, P. Multiple Pathways to Homothallism in Closely Related Yeast Lineages in the Basidiomycota. mBio 2021, 12, 10–128. [Google Scholar] [CrossRef]
- Sopko, R.; Huang, D.; Preston, N.; Chua, G.; Papp, B.; Kafadar, K.; Snyder, M.; Oliver, S.G.; Cyert, M.; Hughes, T.R.; et al. Mapping Pathways and Phenotypes by Systematic Gene Overexpression. Mol. Cell 2006, 21, 319–330. [Google Scholar] [CrossRef]








| Gene Id | Description | WT 518 vs. ter1-02 | WT 518 vs. ter1-24 | ter1-02 vs. ter1-24 |
|---|---|---|---|---|
| UMAG_12032* | Related to ATP-dependent DNA helicase 1, 2 | 3.161 | 5.206 | 2.067 |
| UMAG_12076 u | Related to ATP-dependent DNA helicase 1, 2 | 4.939 | 0.225 | −4.688 |
| UMAG_06476* | Related to RecQ helicase 1, 3 | 4.352 | 0.298 | −4.028 |
| UMAG_0647 * | Related to RecQ helicase 1, 3 | 2.924 | 0.639 | −2.259 |
| UMAG_11065* | Related to RecQ helicase 1, 3 | 6.509 | — | −9.438 |
| UMAG_04094* | Related to RecQ helicase 1, 3 | 8.136 | 0.455 | −7.651 |
| UMAG_04308* | Related to RecQ helicase 1, 3 | 7.013 | 0.308 | −6.679 |
| Gene Id | Description | WT 518 vs. ter1-02 | WT 518 vs. ter1-24 | ter1-02 vs. ter1-24 |
|---|---|---|---|---|
| UMAG_01758 | Related to multidrug resistance-associated protein 1 | 2.756 | 0.040 | −2.692 |
| UMAG_01937 | Related to sphingomyelin phosphodiesterase | 1.003 | 2.116 | 1.136 |
| UMAG_01965 | Related to solute carrier family 40 member 2 | 1.629 | 2.87 | 1.263 |
| UMAG_02224 | Related to palmitoyltransferase ZDHHC16 | −1.678 | 1.149 | 2.851 |
| UMAG_02753 | Related to peroxygenase 2 | 5.517 | — | −3.186 |
| UMAG_02803 | Related to beta-1,3-glucan-binding protein | 7.852 | — | −11.899 |
| UMAG_03073 | Related to glutathione S-transferase 3 | 1.176 | 2.027 | 0.881 |
| UMAG_03122 * | Related to beta-1,3-glucan-binding protein | 2.118 | 0.938 | −1.161 |
| UMAG_03177 | Related to peroxisomal membrane-associated protein 20 | 2.293 | 2.788 | 0.519 |
| UMAG_03728 | Related to indoleamine 2,3-dioxygenase 1 | −0.804 | 1.589 | 2.421 |
| UMAG_03881 | Related to 30 kDa heat shock protein | 4.207 | 1.426 | −2.755 |
| UMAG_04410 | Related to MFS siderochrome iron transporter C | −0.794 | −4.970 | −4.148 |
| UMAG_05600 | Related to succinate-semialdehyde dehydrogenase [NADP(+)] | 2.335 | 1.806 | −0.503 |
| UMAG_06404 | Related to peroxiredoxin PRX1, mitochondrial | 0.775 | −1.938 | −2.688 |
| UMAG_10131 | Related to phosphatidylserine decarboxylase proenzyme 2 | 2.234 | 2.083 | −0.127 |
| UMAG_10781 | Related to disulfide-bond oxidoreductase YfcG | 3.518 | 2.097 | −1.394 |
| UMAG_11944 | Related to glycerol 2-dehydrogenase (NADP(+)) | 1.495 | −0.928 | −2.399 |
| UMAG_12161 | Related to lipase 5 | −0.163 | −2.497 | −2.305 |
| Gene Id | Description | WT 518 vs. ter1-02 | WT 518 vs. ter1-24 | ter1-02 vs. ter1-24 |
|---|---|---|---|---|
| UMAG_01025 | Related to probable transcriptional regulatory protein STB4 | 2.953 | 2.462 | −0.429 |
| UMAG_01456 | Related to regulatory protein CAT8 3 | 0.758 | 2.057 | 1.323 |
| UMAG_01523 | Fox1—related to fork head domain transcription factor Slp1 1 | 4.985 | — | −3.212 |
| UMAG_02835 | Related to conidiophore development regulator abaA 3 | −0.008 | 2.615 | 2.650 |
| UMAG_03296 | Related to Yap and AP-1-like transcription factor napA | 2.751 | 2.691 | −0.026 |
| UMAG_04101 * | Mtf1—related to Myb-related protein A 2 | 5.413 | — | −3.400 |
| UMAG_04778 | Related to transcriptional repressor XBP1 3 | — | 3.538 | 1.560 |
| UMAG_05721 | Srb1—related to putative transcription factor sre2 3, 4 | −1.494 | 1.386 | 2.904 |
| UMAG_06308 | Related to transcription factor RFX4 3 | 0.727 | 2.138 | 1.432 |
| UMAG_12024 | Hdp1—related to short stature homeobox protein 5 | 2.476 | 3.108 | 0.647 |
| UMAG_12304 | Related to positive regulator of purine utilization | 4.330 | 0.912 | −3.390 |
| UMAG_03172 | Rbf1—related to zinc finger protein 2 6 | 0.803 | 2.041 | 1.249 |
| UMAG_02775 | Unh1 7 | 3.315 | 3.281 | −0.011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanpedro-Luna, J.A.; Vega-Alvarado, L.; Vázquez-Cruz, C.; Sánchez-Alonso, P. Global Gene Expression of Post-Senescent Telomerase-Negative ter1Δ Strain of Ustilago maydis. J. Fungi 2023, 9, 896. https://doi.org/10.3390/jof9090896
Sanpedro-Luna JA, Vega-Alvarado L, Vázquez-Cruz C, Sánchez-Alonso P. Global Gene Expression of Post-Senescent Telomerase-Negative ter1Δ Strain of Ustilago maydis. Journal of Fungi. 2023; 9(9):896. https://doi.org/10.3390/jof9090896
Chicago/Turabian StyleSanpedro-Luna, Juan Antonio, Leticia Vega-Alvarado, Candelario Vázquez-Cruz, and Patricia Sánchez-Alonso. 2023. "Global Gene Expression of Post-Senescent Telomerase-Negative ter1Δ Strain of Ustilago maydis" Journal of Fungi 9, no. 9: 896. https://doi.org/10.3390/jof9090896