3.2. Genome Sequencing and Quality Assessment
To present sequencing quality, the sequencing metrics of the two lichen samples HPH and HTU are listed in
Table 2. Obtained metrics are comparable in each sample with only slight deviations. Due to mean HiFi Read Quality well above Q20, all samples were eligible for further bioinformatic analyses.
After taxonomic evaluation of both lichen samples, bins on family and genus level representing the different taxonomic groups of the investigated
Hypogymnia holobionts were selected, in order to provide a sufficient insight on community composition.
Table 3 shows the chosen bins for HPH and HTU divided into eukaryotic and bacterial origin. The identified fungi belonging to
Herpotrichiellaceae and
Pleosporineae are lichen-associated Ascomycota which possibly belong to the so-called “black fungi” (
Eurotiomycetes and
Dothideomycetes) [
78].
To evaluate genome completeness and reliability for further data processing, a BUSCO [
66] assessment was performed on the respective bins of the lichen metagenome. In both samples, the bin “
Parmeliaceae” included sequences of the mycobiont and the bin “
Trebouxia” sequences of the photobiont. Unfortunately, the amount of lichen genomes available in databases is too low to provide a selection on lower taxonomic level. Bin compositions mainly deviated in regard to
Herpotrichiellaceae, which was not prominent in HTU, whereas the bin of
Verrucomicrobiota was not as prevalent in HPH as the other investigated bins.
Subsequently, the metagenomic bins were compared to the respective BUSCO gene sets of
Ascomycota,
Chlorophyta and
Bacteria. These taxonomic groups were selected to provide a reliable insight in regard to genome representation of the lichen community. Obtained BUSCO results are shown in percentages to allow for an accurate comparison between the investigated bins.
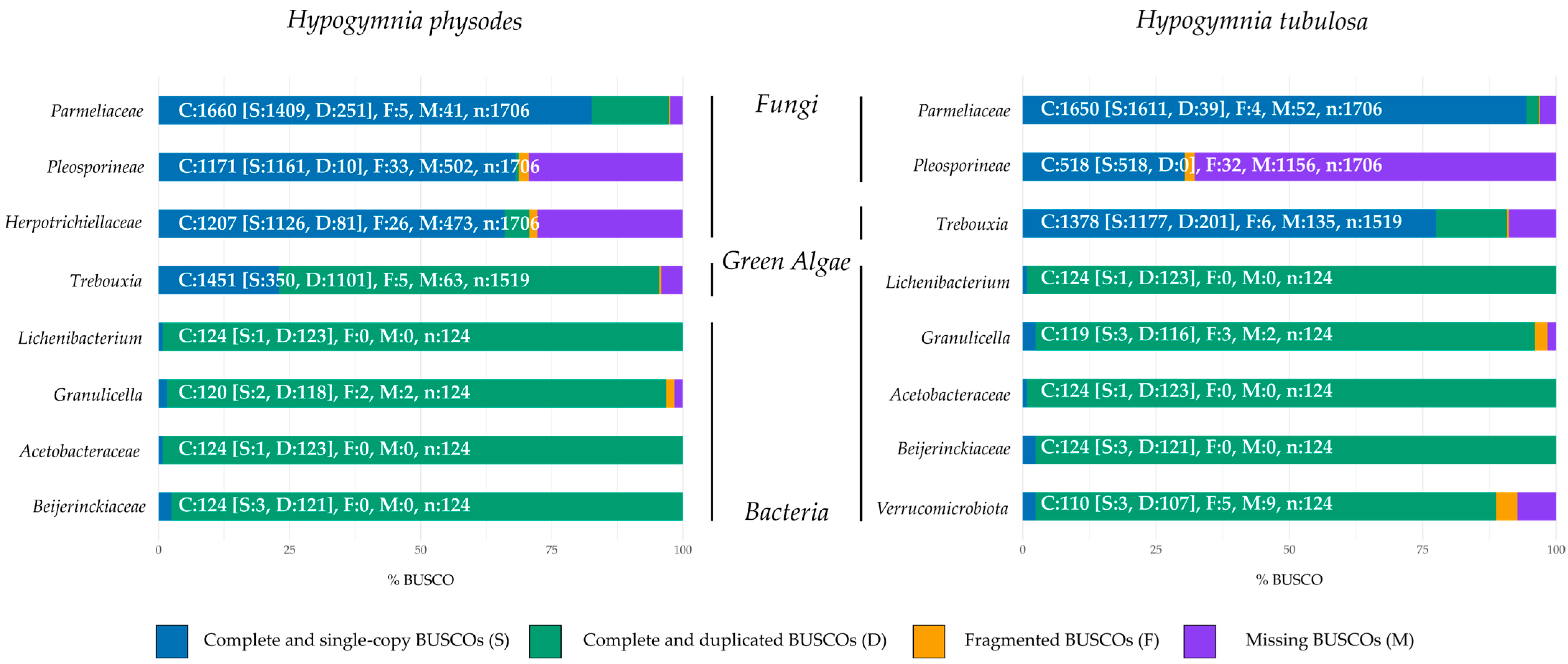
Figure 2 depicts the normalized and summarized results for each lichen sample by respective bin. All columns also include the absolute numbers to allow for more precise comparison between different bins.
In order to evaluate genome completeness of the mycobiont and other fungal bins, the orthologous gene sets of the phylum
Ascomycota odb10 were chosen. In HPH, the fungal bins contained predominantly complete and single BUSCOs (66–82.6%) with the highest values in
Parmeliaceae. Duplicated genes were observed in all fungal bins ranging from 0.6 to 14.7%, increasing from
Pleosporineae over
Herpotrichiellaceae to
Parmeliaceae. These findings are based on fungal diversity in lichen, as the presence of different fungal genomes results in multiple detections of investigated gene sets by BUSCO [
3]. In regard to missing BUSCO gene sets, approximately 29% were absent in
Herpotrichiellaceae and
Pleosporineae. As the primary mycobiont makes up most of the lichen biomass, this genome is predominately sequenced compared to other fungi included in the metagenome. Fragmentation on BUSCO gene sets ranged from 0.3 to 1.9%.
The photobiont (Trebouxia) was compared to the gene set of Chlorophyta odb10, which yielded mainly complete and duplicated BUSCOs (72.5%), with complete and single BUSCOs making up the majority of the remaining gene sets (23%). These findings are in line with the previously elaborated influence of biomass distribution in lichens on sequencing depth. The share of missing BUSCOs was about 4%, whereas 0.3% were fragmented.
BUSCO evaluation of the bacterial factions of the investigated lichen community against the gene set
Bacteria yielded high duplication rates in every bin (95–99.2%). As the investigated bins encased more than one bacterial species, duplication rates were inevitable [
79]. By investigating the genome completeness of the fungal bins present in HTU, the
Parmeliaceae bin exhibits mainly complete and single BUSCOs with only slight duplication or fragmentation. In contrast, a high rate of missing BUSCOs was seen in the bin containing
Pleosporineae. The node of this bin was smaller in comparison to the one in HPH (see
Table S2). These findings may also be based on the share of primary mycobiont present in the lichen sample.
Comparable results are observed in the photobiont bin. Here the duplication rate is considerably lower than in the respective bin of HPH. These findings may suggest a uniform composition of algal partners in this lichen sample and a more diverse composition of algal strains in HPH. The bacterial faction of HTU displayed comparable results to HPH regarding duplication rates, based on the same rationale. The assessment of gene set completeness showed a similar pattern to that of genome completeness across all taxonomic groups belonging to eukaryota (
Supplementary Figure S1). For bacterial bins, more complete and singe BUSCOs were observed by analysis of gene set completeness. However, the rate of missing and fragmented BUSCOs increased.
A second approach to evaluate the contiguity of the assembled metagenomes involves the utilization of the quality assessment tool for genome assemblies (QUAST). Thus, the different bins were assessed based on their genome size, number of contigs, and N50 values; a summary of the statistics for each bin are provided in
Table 4 and
Table 5. Notably, the N50 values for the primary symbionts,
Parmeliacea sp. and
Trebouxia sp., were around 1 Mb for both lichen samples. These findings, combined with the low L50 value for each, suggest with a high degree of confidence, that the genomes are assembled contiguously. A 1.4-fold difference in total contig length of
Parmeliacea is observed between HPH and HTU, which could be due to natural variations in genome size. However, both bins align with the average size of the assembled
Parmelia spp. genome reported in the literature (45 Mb, NCBI BioSample: SAMN17391792). A 1.6-fold difference in genome size can be seen in the
Trebouxia bins; here, it is worth mentioning that in HTU, the missing BUSCOs differ by a factor of two. This may cause variations in observed total contig length. Deviation from the 69.35 Mb genome size (NCBI BioSample: SAMD00066476 [
80]) reported in the literature may be explained by the nature of the
Trebouxia bin, as it possibly encloses more than one algal partner in HPH [
81].
The bacterial bins in HPH and HTU exhibit a high number of contigs of up to 2647. However, it should be noted that the reported genome sizes for
Lichenibacterium,
Acetobacteraceae, and
Granulicella are around 6 Mb (NCBI BioSample: SAMN09781801), 3 Mb (NCBI BioSample: SAMN29020633), and 6 Mb (NCBI BioSample: SAMN28407668, [
82]), respectively. Usually, bacterial genomes processed by long read sequencers are expected to be one circular contig only. This suggests that multiple organisms belonging to these genera may be present in a single bin. Additionally, the high duplication rates observed in all bacterial bins by the above BUSCO data is consistent with the high number of contigs and contig lengths found. By investigating the average coverage in both lichen samples, a strong bias towards the mycobiont is observed, 251- and 284-fold coverage in HPH and HTU, respectively. These findings are in line with the aforementioned distribution of mycobiont biomass in lichen. The algal and other fungal partners in both samples are comparable in coverage. As the bacterial bins of HPH and HTU differed in size, it was also expected to be observed in regard to coverage.
Comparison of GC content yields similar results throughout all compared taxonomic groups in both lichen samples.
3.3. Gene Models and Functional Annotation
Annotation of gene models was conducted with AUGUSTUS, utilizing the presented metagenome assemblies and a long-read IsoSeq database functioning as hints [
70,
71,
72]. Here transcriptomic data from each lichen sample was deployed as an individual training set to ensure correct gene predictions for all factions of the respective metagenome [
83]. For both lichen samples, the predicted genes resembling putative proteins were summed up from all encased bins investigated in this study. This yielded a total gene count of 133,963 for HPH and 96,257 for HTU. In addition, statistics on average genes length, gene density, and introns per gene were evaluated to further validate obtained gene predictions. Exact numbers for each bin are depicted in
Table 6.
To further elaborate the metagenomes, a functional annotation was conducted based on the databases of Cluster of Orthologous Groups (COG) and Gene Ontology (GO) terms. Results were similar in both lichen samples and their respective bins. Annotation rates differed from ~85% in Parmeliaceae to ~94% in Trebouxia regarding the eukaryotic fraction, whereas approximately ~93% in the bacterial bins could be annotated. Interestingly, the bins of Parmeliaceae and Trebouxia harbored ~26% and ~20% of predicted genes with no assigned function, making further research on these organisms highly interesting.
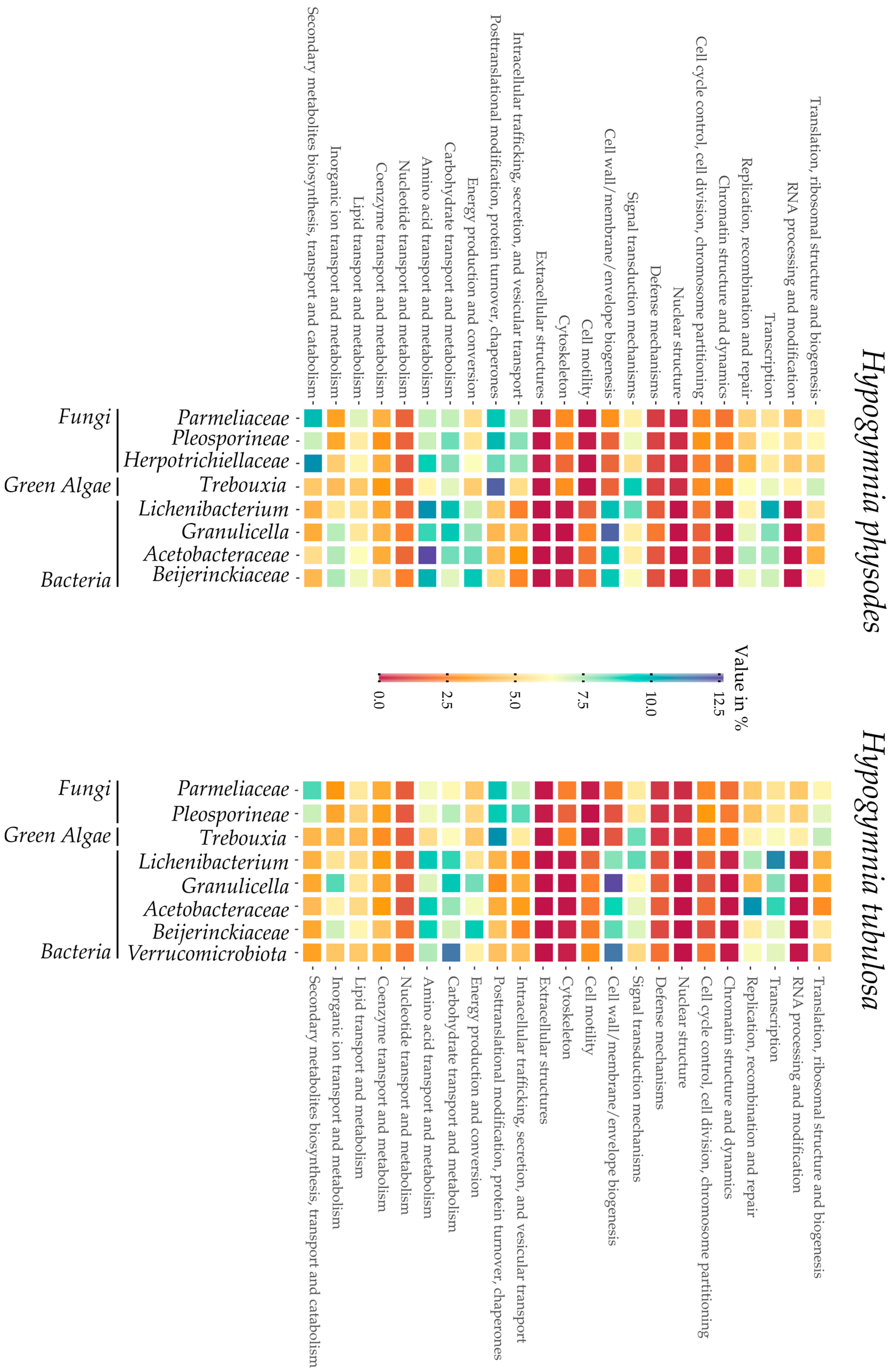
Visualization of the COG distribution in the investigated lichen samples by each bin is depicted in
Figure 3. From left to right, the bins are sorted by taxonomic group, beginning with the fungal fraction of the lichen metagenome over the green algae and ending with the bacterial bins. This overview provides insights into the respective functionalities of the predicted genes in each bin. Furthermore, strong differences between eukaryotes and prokaryotes are highlighted by a deep red or blue coloring in e.g., chromatin structure or cell wall biogenesis. On first sight, both heatmaps display a comparable coloring in the respective bins, thus highlighting the high similarity of the samples HPH and HTU. In regard to biosynthetic potential of the
Hypogymnia holobiont, the row dedicated to “secondary metabolite biosynthesis” is elaborated further. Intriguingly, both
Parmeliaceae bins possess high amounts of proteins predicted to be involved in secondary metabolite production. Other fungal bins equally displayed increased values compared to the bacterial faction or the photobiont in the same category. These findings are coherent with the observation of enhanced secondary metabolite production in lichen mycobionts and fungi [
17,
84,
85,
86,
87].
3.4. Biosynthetic Gene Clusters
Since the genus
Hypogymnia is known to produce a manifold secondary metabolite spectrum, a deeper investigation into the biosynthetic pathways may yield intriguing results [
8,
88,
89]. Therefore, the here-presented metagenomes were evaluated for biosynthetic gene clusters (BGCs) by antiSMASH 6.1.1 fungal/bacterial version [
73], based on the previously conducted gene annotation. Furthermore, both
Trebouxia bins were annotated by the fungal version of antiSMASH because plantiSMASH yielded insufficient results as previously described [
90]. With this in silico approach, the biosynthetic enzymes involved in the formation of unique lichen substances are rendered accessible for wet lab research. To visualize the biosynthetic potential of the genus
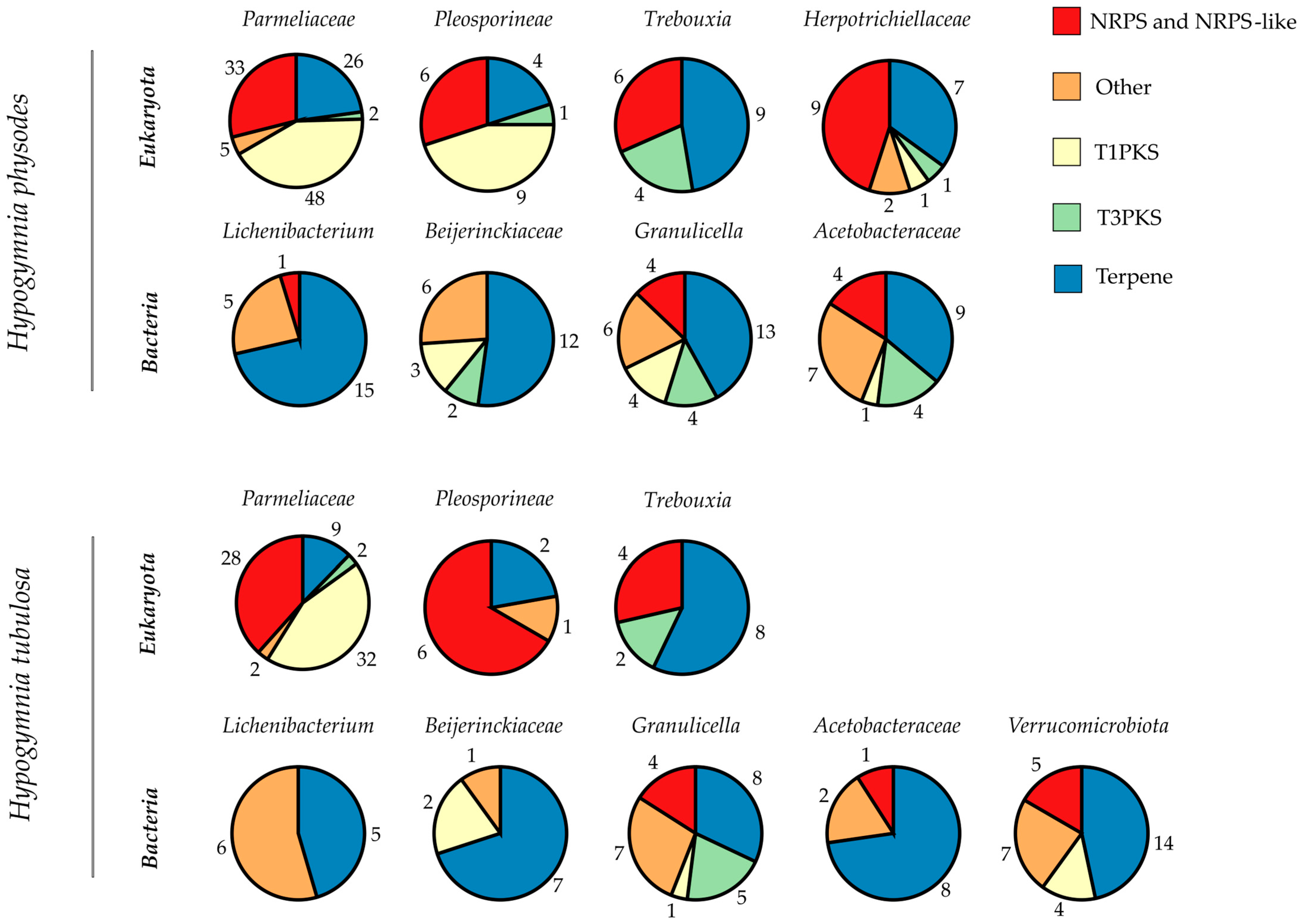
Hypogymnia, each bin’s BGC composition was categorized by putative function (
Figure 4). Bins are grouped according to lichen sample and subdivided by superkingdom. The pie charts were computed based on total numbers of BGCs in the respective bin, including numbers representing the amount of BGCs of the various types. Main BGC categories identified by antiSMASH were nonribosomal peptide synthetases (NRPS and NRPS-like), type I and III polyketide synthases (T1PKS, T3PKS), and terpenes. All remaining types were grouped as “Other”. The complete list of all identified BGCs is given in
Tables S3 and S4.
Most BGCs annotated throughout both lichen samples were related to terpene formation, followed by decreasing numbers of NRPS over T1PKS and ending with T3PKS. To allow for a direct comparison, the total numbers of BGCs per bin are listed in
Table 7. For the first time, the photobiont and the bacterial factions were investigated directly from a lichen thallus. This gives information on the BGC composition of the complete holobiont as found in nature.
The prokaryotic faction in HPH exhibited an average of 26 BGCs over all four bins. For HTU, an average of ~17 BGCs was reported. Some of the bacterial bins exhibit BGC totals in the ranges comparable to
Streptomyces strains, which are known for their multitude of BGCs (25–70) [
91]. However, as the bins contain more than one bacterial genome the exact numbers of BGCs may differ between individual species. Main BGC class assigned by antiSMASH was related to terpene formation. Throughout all bacterial bins, only 26 BGCs were with assigned function. The influence of lichen-associated bacteria on the symbiosis still remains mostly unclear [
92]. Identification of bacterial BGCs may yield insights on further functions in addition to the already known provision of vitamins and cofactors used for degradation of phenolic compounds [
93]. As most of the BGCs remain without assigned function, these may be intriguing for further research.
A closer look into the photobiont bins yields a comparable distribution of BGCs in the respective categories, even if the BUSCO and QUAST results differed strongly (see
Figure 2 and
Table 4 and
Table 5). For the algal bins, 19 (HPH) and 14 (HTU) BGCs were annotated which exceeds the numbers reported in previous studies [
90]. This might be due to the nature of the investigated bins, enclosing more than one species. Therefore, an ITSx analysis was performed to elucidate the bin composition of
Trebouxia and
Parmeliaceae. For the bins in HPH only single ITS sequences were found, which is also true for the mycobiont bin in HTU. In regard to the photobiont bin in HTU, no ITS sequence was extractable. However, the absence of ITS sequences does not eliminate the possibility that other parts of the respective genome are present in the respective bins. This also applies to the findings of the BUSCO analysis. Results on ITSx are listed in
Table S5. As algae from the genus
Trebouxia exhibit slow growth rates or are often not yet amenable for cultivation [
43], this metagenomic approach renders unidentified BGCs and genes accessible for genome mining. Intriguingly, antiSMASH yielded no assignment to any of the annotated BGCs, making these clusters interesting for further research. As the main class of photobiont BGCs is related to terpene production a formation of relevant natural products may be possible.
In comparison, the
Parmeliaceae bins harbor the highest number of BGCs (114 in HPH; 73 in HTU) in the respective lichen. The richness in BGCs is in line with findings in previous studies concerning HTU, as the described BGC content in lichen ranges between 27 and 80 [
10]. In the case of HPH, the total number of BGCs exceeded these amounts. Compared to other organisms harboring high amounts of BGCs such as
Streptomyces spp. (23–80) [
94,
95],
Cyanobacteria spp. (1–42) [
96,
97]
Myxobacteria spp. (30–46) [
98,
99], and
Nocardia spp. (~36) [
100,
101], the investigated
Parmeliaceae bins showed comparable or higher totals of BGCs. For both mycobiont bins, the majority of BGCs were putative PKS followed by terpene related clusters. In regard to annotation by antiSMASH with the MIBiG database and Pfam, only 27 (HPH) and 16 (HTU) BGCs were with assigned function, highlighting the yet still untapped potential for further investigation.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}