
Hybrid De Novo Whole-Genome Assembly, Annotation, and Identification of Secondary Metabolite Gene Clusters in the Ex-Type Strain of Chrysosporium keratinophilum

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Sequencing and Assembling
2.2. Genomic Indexes
2.3. Genome Prediction and Annotation of the Strain CBS 104.62
3. Results and Discussion
3.1. Genome Information and Comparison with the Closest Species
3.2. Average Nucleotide Identity
3.3. Prediction of Genes from the Assembled Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sturm, J. Deutschlands Flora 3; Nabu Press: Nurnberg, Germany, 1833. [Google Scholar]
- Saccardo, P.A. Sylloge Fungorum; Octave Doin Edidit: Paris, France, 1901; Volume 15. [Google Scholar]
- Hughes, S.J. Revisiones hyphomycetum aliquot cum appendice de nominibus rejiciendis. Can. J. Bot. 1958, 36, 727–836. [Google Scholar] [CrossRef]
- Carmichael, J.W. Chrysosporium and some other aleuriosporic hyphomycetes. Can. J. Bot. 1962, 40, 1137–1173. [Google Scholar] [CrossRef]
- Dominik, T. Chrysosporium Corda; Zesz nauk wyzsz Szk roln Szczec: Szczecin, Poland, 1967; Volume 24. [Google Scholar]
- Oorschot, V. A revision of Chrysosporium and allied genera. Stud. Mycol. 1980, 20, 1–89. [Google Scholar]
- Kendrick, W.B. The Whole Fungus; National Museums of Canada, Kananaskis Foundation: Ottawa, ON, Canada, 1979. [Google Scholar]
- Dongyou, L. Molecular Detection of Human Fungal Pathogens, 1st ed.; Liu, D., Ed.; CRC Press: Boca Raton, FL, USA, 2011; ISBN 9780429134630. [Google Scholar]
- Stchigel, A.M.; Sutton, D.A.; Cano-Lira, J.F.; Cabañes, F.J.; Abarca, L.; Tintelnot, K.; Wickes, B.L.; García, D.; Guarro, J. Phylogeny of chrysosporia infecting reptiles: Proposal of the new family Nannizziopsiaceae and five new species. Pers. Mol. Phylogeny Evol. Fungi 2013, 31, 86–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal, P.; Vinuesa, M.A.; Sanchez-Puelles, J.A.; Guarro, J. Phylogeny of the anamorphic genus Chrysosporium and related taxa based on rDNA internal transcribed spacer sequences. In Kushwaha RKS; Guarro, J., Ed.; Biology of Dermatophytes and Other Keratinophilic Fungi; Rev. Iberoam. Micol.: Bilbao, Spain, 2000; pp. 22–28. [Google Scholar]
- Sigler, L. Ascomycetes: The Onygenaceae and other fungi from the order Onygenales. In Pathogenic Fungi in Humans and Animals; Marcel Dekker, Inc.: New York, NY, USA, 2002; pp. 195–236. ISBN 0824706. [Google Scholar]
- Palma, M.Á.G.; Espín, L.Á.; Pérez, A.F.; León, J.Á.M. Invasive Sinusal Mycosis due to Chrysosporium tropicum. Acta Otorrinolaringol. (Engl. Ed.) 2007, 58, 164–166. [Google Scholar] [CrossRef]
- Maruthi, Y.A.; Lakshmi, K.A.; Rao, S.R.; Hossain, K.; Chaitanya, D.A.; Karuna, K. Dermatophytes and other fungi associated with hair-scalp of Primary school children in Visakhapatnam, India: A Case Study And Literature Review. Internet J. Microbiol. 2008, 5, 1–4. [Google Scholar]
- Mijiti, J.; Pan, B.; de Hoog, S.; Horie, Y.; Matsuzawa, T.; Yilifan, Y.; Liu, Y.; Abliz, P.; Pan, W.; Deng, D.; et al. Severe chromoblastomycosis-like cutaneous infection caused by Chrysosporium keratinophilum. Front. Microbiol. 2017, 8, 83. [Google Scholar] [CrossRef] [Green Version]
- Calvo, R.M.; Calvo, M.A.; Larrondo, J. Enzyme activities in Chrysosporium strains. Mycopathologia 1991, 116, 177–179. [Google Scholar] [CrossRef]
- Hopsu-Havu, V.K.; Sonck, C.E.; Tunnela, E. Production of Elastase by Pathogenic and Non-Pathogenic Fungi. Mycoses 1972, 15, 105–110. [Google Scholar] [CrossRef]
- Slater, G.P.; Haskins, R.H.; Hogge, L.R. Metabolites from a Chrysosporium species. Can. J. Microbiol. 1971, 17, 1576–1579. [Google Scholar] [CrossRef]
- Singh, C.J. Optimization of an extracellular protease of Chrysosporium keratinophilum and its potential in bioremediation of keratinic wastes. Mycopathologia 2003, 156, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ganaie, M.A.; Sood, S.; Rizvi, G.; Khan, T.A. Isolation and Identification of Keratinophilic Fungi from Different Soil Samples in Jhansi City (India). Plant Pathol. J. 2010, 9, 194–197. [Google Scholar] [CrossRef] [Green Version]
- Bohacz, J.; Korniłłowicz-Kowalska, T.; Kitowski, I.; Ciesielska, A. Degradation of chicken feathers by Aphanoascus keratinophilus and Chrysosporium tropicum strains from pellets of predatory birds and its practical aspect. Int. Biodeterior. Biodegrad. 2020, 151, 104968. [Google Scholar] [CrossRef]
- Frey, D.; Carmichael, J.W. Chrysosporium keratinophilum. Can. J. Bot. 1962, 40, 1157. [Google Scholar] [CrossRef]
- Cano, J.; Guarro, J. The genus Aphanoascus. Mycol. Res. 1990, 94, 355–377. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 8 July 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Puiu, D.; Luo, M.-C.; Zhu, T.; Koren, S.; Marçais, G.; Yorke, J.A.; Dvořák, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, R.M.; Seppey, M.; Sim, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M.; Rosenberg, M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2015, 8, 12–24. [Google Scholar] [CrossRef]
- Seemann, T. Barrnap. Available online: https://github.com/tseemann/barrnap (accessed on 8 July 2022).
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinforma 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Seager, J.; Wood, V.; Rutherford, K.; Venkatesh, S.Y.; De Silva, N.; Martinez, M.C.; Pedro, H.; Yates, A.D.; et al. PHI-base: The pathogen–host interactions database. Nucleic Acids Res. 2020, 48, D613–D620. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Wibberg, D.; Stadler, M.; Lambert, C.; Bunk, B.; Spröer, C.; Rückert, C.; Kalinowski, J.; Cox, R.J.; Kuhnert, E. High quality genome sequences of thirteen Hypoxylaceae (Ascomycota) strengthen the phylogenetic family backbone and enable the discovery of new taxa. Fungal Divers. 2021, 106, 7–28. [Google Scholar] [CrossRef]
- Sharpton, T.J.; Stajich, J.E.; Rounsley, S.D.; Gardner, M.J.; Wortman, J.R.; Jordar, V.S.; Maiti, R.; Kodira, C.D.; Neafsey, D.E.; Zeng, Q.; et al. Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res. 2009, 19, 1722–1731. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, C.A.; Champion, M.D.; Holder, J.W.; Muszewska, A.; Goldberg, J.; Bailão, A.M.; Brigido, M.M.; da Silva Ferreira, M.E.; Garcia, A.M.; Grynberg, M.; et al. Comparative genomic analysis of human fungal pathogens causing paracoccidioidomycosis. PLoS Genet. 2011, 7, e1002345. [Google Scholar] [CrossRef] [Green Version]
- Whiston, E.; Taylor, J.W. Comparative phylogenomics of pathogenic and nonpathogenic species. G3 Genes Genomes Genet. 2016, 6, 235–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persinoti, G.F.; Martinez, D.A.; Li, W.; Döğen, A.; Billmyre, R.B.; Averette, A.; Goldberg, J.M.; Shea, T.; Young, S.; Zeng, Q.; et al. Whole-genome analysis illustrates global clonal population structure of the ubiquitous dermatophyte pathogen Trichophyton rubrum. Genetics 2018, 208, 1657–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, L.; Bitencourt, T.A.; Lang, E.A.S.; Sanches, P.R.; Peres, N.T.A.; Rossi, A.; Martinez-Rossi, N.M. Genes coding for LysM domains in the dermatophyte Trichophyton rubrum: A transcription analysis. Med. Mycol. 2020, 58, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Kothari, D.; Rani, A.; Goyal, A. Keratinases. In Current Developments in Biotechnology and Bioengineering. Production, Isolation and Purification of Industrial Products; Elsevier: Amsterdam, The Netherlands, 2017; pp. 447–469. [Google Scholar] [CrossRef]
- Qiu, J.; Wilkens, C.; Barrett, K.; Meyer, A.S. Microbial enzymes catalyzing keratin degradation: Classification, structure, function. Biotechnol. Adv. 2020, 44, 107607. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Jin, H.S.; La, J.W.; Sung, J.Y.; Park, S.Y.; Kim, W.C.; Lee, D.W. Identification of keratinases from Fervidobacterium islandicum AW-1 using dynamic gene expression profiling. Microb. Biotechnol. 2020, 13, 442–457. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.P.D.O.; Cai, G.; Nachtschatt, M.; Navone, L.; Zhang, Z.; Robins, K.; Speight, R. Challenges and Opportunities in Identifying and Characterising Keratinases for Value-Added Peptide Production. Catalysts 2020, 10, 184. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiei, V.; Vélëz, H.; Tzelepis, G. The Role of Glycoside Hydrolases in Phytopathogenic Fungi and Oomycetes Virulence. Int. J. Mol. Sci. 2021, 22, 9359. [Google Scholar] [CrossRef]
- Mitchell, N.M.; Grys, T.E.; Lake, D.F. Carbo-loading in Coccidioides spp.: A quantitative analysis of CAZyme abundance and resulting glycan populations. Glycobiology 2020, 30, 186–197. [Google Scholar] [CrossRef]
- Park, Y.J.; Jeong, Y.U.; Kong, W.S. Genome Sequencing and Carbohydrate-Active Enzyme (CAZyme) Repertoire of the White Rot Fungus Flammulina elastica. Int. J. Mol. Sci. 2018, 19, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Kim, S.L.; Jeong, M.-K.; Yu, O.H.; Eyun, S.; Nguyen, H.; Choi, H.; Kim, S.L.; Jeong, M.-K.; Yu, O.H.; et al. Identification and Phylogenetic Analysis of Chitin Synthase Genes from the Deep-Sea Polychaete Branchipolynoe onnuriensis Genome. J. Mar. Sci. Eng. 2022, 10, 598. [Google Scholar] [CrossRef]
- Yonekura-Sakakibara, K.; Hanada, K. An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 2011, 66, 182–193. [Google Scholar] [CrossRef]
- Díaz-Jiménez, D.F. Fungal Mannosyltransferases as Fitness Attributes and their Contribution to Virulence. Curr. Protein Pept. Sci. 2017, 18, 1065–1073. [Google Scholar] [CrossRef]
- Salam, L.B. Detection of carbohydrate-active enzymes and genes in a spent engine oil-perturbed agricultural soil. Bull. Natl. Res. Cent. 2018, 42, 10. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Gong, W.; Zhu, Z.; Yan, L.; Hu, Z.; Peng, Y. Comparative transcriptomics of Pleurotus eryngii reveals blue-light regulation of carbohydrate-active enzymes (CAZymes) expression at primordium differentiated into fruiting body stage. Genomics 2018, 110, 201–209. [Google Scholar] [CrossRef]
- Agrawal, Y.; Narwani, T.; Subramanian, S. Genome sequence and comparative analysis of clavicipitaceous insect-pathogenic fungus Aschersonia badia with Metarhizium spp. BMC Genom. 2016, 17, 367. [Google Scholar] [CrossRef] [Green Version]
- Hage, H.; Rosso, M.N. Evolution of Fungal Carbohydrate-Active Enzyme Portfolios and Adaptation to Plant Cell-Wall Polymers. J. Fungi 2021, 7, 185. [Google Scholar] [CrossRef]
- Sützl, L.; Laurent, C.V.F.P.; Abrera, A.T.; Schütz, G.; Ludwig, R.; Haltrich, D. Multiplicity of enzymatic functions in the CAZy AA3 family. Appl. Microbiol. Biotechnol. 2018, 102, 2477–2492. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.M.; de Almeida, L.G.; Kubitschek-Barreira, P.; Alves, F.L.; Kioshima, É.S.; Abadio, A.K.; Fernandes, L.; Derengowski, L.S.; Ferreira, K.S.; Souza, R.C.; et al. Comparative genomics of the major fungal agents of human and animal Sporotrichosis: Sporothrix schenckii and Sporothrix brasiliensis. BMC Genom. 2014, 15, 943. [Google Scholar] [CrossRef] [Green Version]
- Petrasch, S.; Silva, C.J.; Mesquida-Pesci, S.D.; Gallegos, K.; van den Abeele, C.; Papin, V.; Fernandez-Acero, F.J.; Knapp, S.J.; Blanco-Ulate, B. Infection Strategies Deployed by Botrytis cinerea, Fusarium acuminatum, and Rhizopus stolonifer as a Function of Tomato Fruit Ripening Stage. Front. Plant Sci. 2019, 10, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitowski, I.; Korniłłowicz-Kowalska, T.; Bohacz, J.; Ciesielska, A. Dispersal of Aphanoascus keratinophilus by the rook Corvus frugilegus during breeding in East Poland. Sci. Rep. 2022, 12, 2142. [Google Scholar] [CrossRef] [PubMed]
- Hagee, D.; Abu Hardan, A.; Botero, J.; Arnone, J.T. Genomic clustering within functionally related gene families in Ascomycota fungi. Comput. Struct. Biotechnol. J. 2020, 18, 3267–3277. [Google Scholar] [CrossRef]
- Izumi, Y.; Ohtani, K.; Miyamoto, Y.; Masunaka, A.; Fukumoto, T.; Gomi, K.; Tada, Y.; Ichimura, K.; Peever, T.L.; Akimitsu, K. A Polyketide Synthase Gene, ACRTS2, Is Responsible for Biosynthesis of Host-Selective ACR-Toxin in the Rough Lemon Pathotype of Alternaria alternata. Mol. Plant-Microbe Interact. 2012, 25, 1419–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godio, R.P.; Fouces, R.; Gudiña, E.J.; Martín, J.F. Agrobacterium tumefaciens-mediated transformation of the antitumor clavaric acid-producing basidiomycete Hypholoma sublateritium. Curr. Genet. 2004, 46, 287–294. [Google Scholar] [CrossRef]
- Chen, L.H.; Yang, S.L.; Chung, K.R. Resistance to oxidative stress via regulating siderophore-mediated iron acquisition by the citrus fungal pathogen Alternaria alternata. Microbiology 2014, 160, 970–979. [Google Scholar] [CrossRef]
- Swift, C.L.; Louie, K.B.; Bowen, B.P.; Olson, H.M.; Purvine, S.O.; Salamov, A.; Mondo, S.J.; Solomon, K.V.; Wright, A.T.; Northen, T.R.; et al. Anaerobic gut fungi are an untapped reservoir of natural products. Proc. Natl. Acad. Sci. USA 2021, 118, e2019855118. [Google Scholar] [CrossRef]






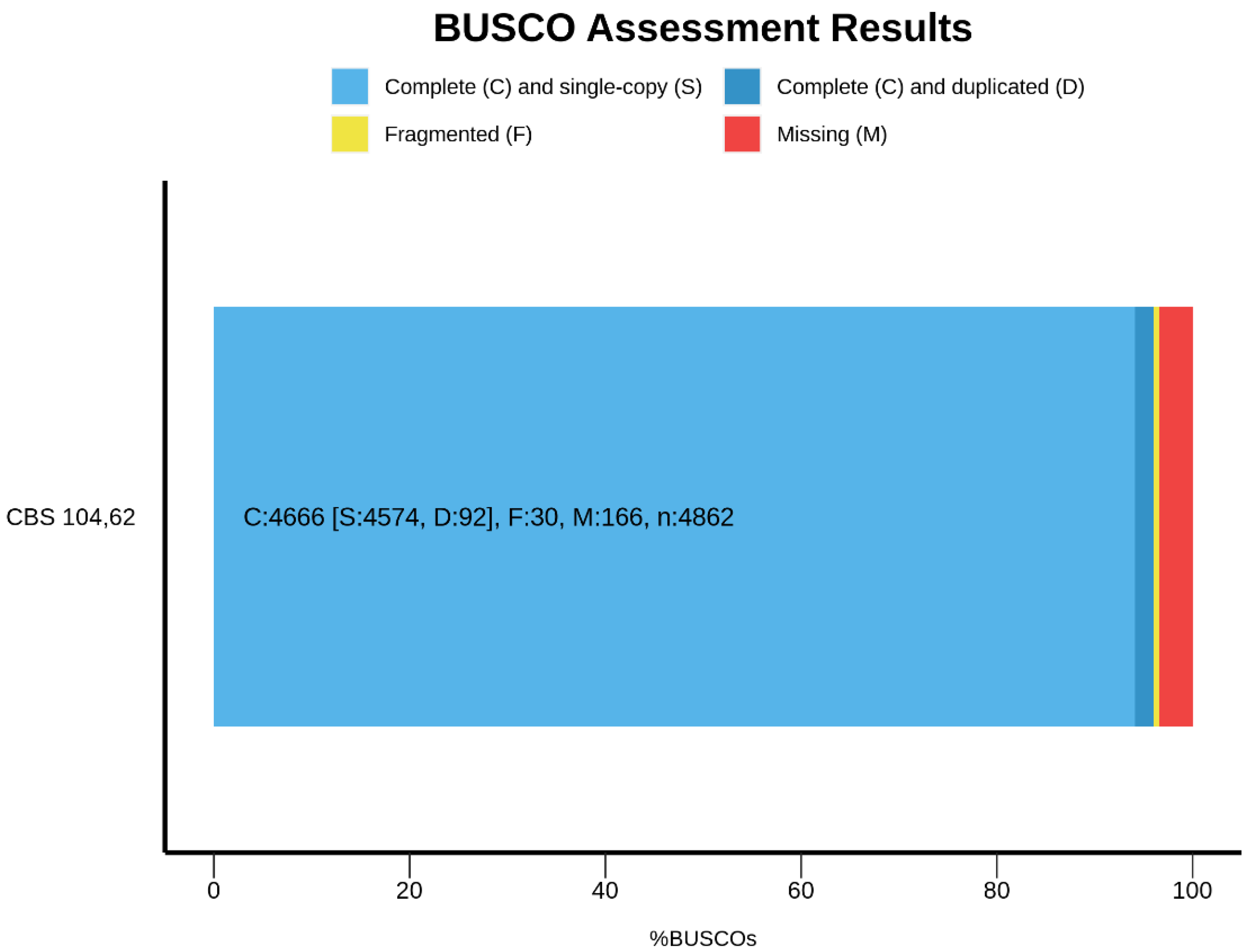{kind=link}
{kind=link}
{kind=link}
{kind=link}
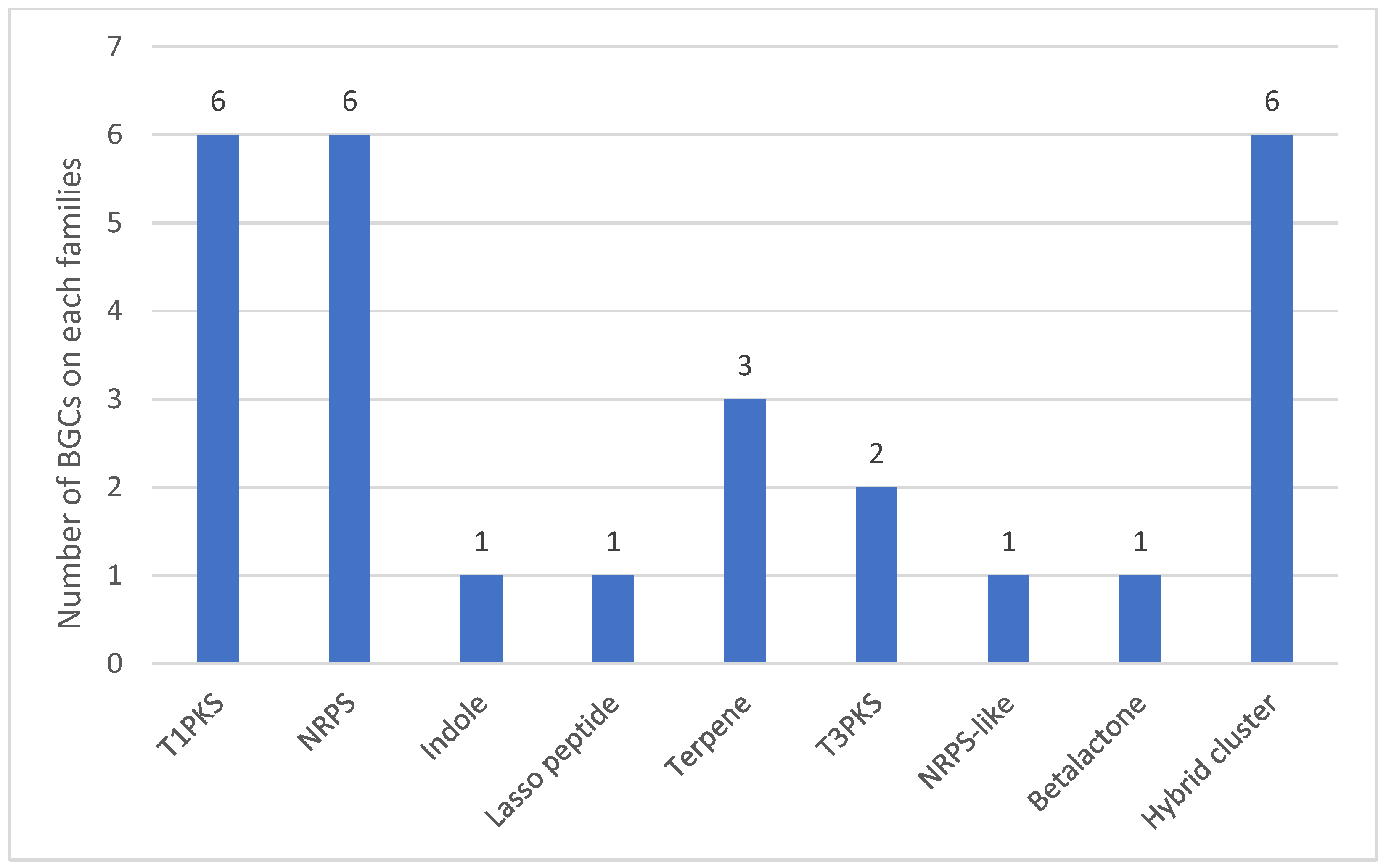{kind=link}
| No. | Species | Strain | GenBank Strain BioSample * | GenBank Assembly Accession | Level | Release Date |
|---|---|---|---|---|---|---|
| 1 | Amauroascus niger (asexual morph unknown) | UAMH 3544 | SAMN03741936 | GCA_001430945.1 | Scaffold | 22/04/22 |
| 2 | Aphanoascus verrucosus (asexual morph Chrysosporium tropicum) | IHEM 4434 | SAMN15800566 | GCA_014839905.1 | Scaffold | 22/04/22 |
| 3 | Brunneospora queenslandica (asexual morph Chrysosporium queenslandicum) | CBS 280.77 | SAMN03741938 | GCA_001430955.1 | Scaffold | 22/04/22 |
| 4 | Coccidioides immitis (asexual morph malbranchea-like) | RS | SAMN02953601 | GCA_000149335.2 | Scaffold | 24/04/22 |
| 5 | Coccidioides posadasii (asexual morph malbranchea-like) | C735 delta SOWgp | SAMN02953748 | GCA_000151335.1 | Scaffold | 24/04/22 |
| 6 | Ophidiomyces ophidiicola (asexual morph chrysosporium-like) | CBS 122913 | SAMN23192527 | GCA_022830035.1 | Scaffold | 22/04/22 |
| 7 | Uncinocarpus reesii (asexual morph unknown) | UAMH 1704 | SAMN02953631 | GCA_000003515.2 | Scaffold | 22/04/22 |
| Genome Statistics | Chrysosporium keratinophilum CBS 104.62T | Aphanoascus verrucosus (Asexual Morph Chrysosporium tropicum) IHEM 4434T | Brunneospora queenslandica (Asexual Morph Chrysosporium queenslandicum) CBS 280.77T | Ophidiomyces ophidiicola (Asexual Morph Chrysosporium-like) CBS 122913T | Uncinocarpus reesii (Asexual Morph Unknown) UAMH 1704 | Coccidioides immitis (Asexual Morph Malbranchea-like) RS | Coccidioides posadasii (Asexual Morph Malbranchea-like) C735 Delta SOWgp |
|---|---|---|---|---|---|---|---|
| Contigs (≥0 bp) | 25 | 211 | 2724 | 116 | 45 | 7 | 55 |
| Total length (≥0 bp) | 25,439,844 | 23,059,040 | 32,335,957 | 21,970,319 | 22,349,738 | 29,016,019 | 27,013,412 |
| Largest contig (bp) | 5,001,415 | 894,230 | 979,930 | 1,803,704 | 7,891,746 | 8,482,323 | 5,398,309 |
| G+C content (%) | 49.09 | 49.59 | 53.15 | 47.64 | 48.66 | 45.96 | 46.59 |
| N50 (bp) | 2,037,736 | 431,852 | 173,991 | 506,472 | 5,332,914 | 4,323,945 | 2,376,830 |
| N90 (bp) | 460,884 | 132,993 | 4367 | 122,144 | 2,507,206 | 3,458,857 | 974,251 |
| L50 | 4 | 17 | 47 | 12 | 2 | 3 | 4 |
| L90 | 14 | 52 | 798 | 42 | 5 | 6 | 11 |
| # N’s per 100 kbp | 10.06 | 10.51 | 6001.89 | 1.81 | 812.87 | 1.38 | 0.12 |
| Carbohydrate-Active Enzyme (CAZyme) Classes | Number of Identified Families | Number of Identified Genes |
|---|---|---|
| Glycoside hydrolases (GHs) | 61 | 156 |
| Glycosyltransferases (GTs) | 41 | 151 |
| Carbohydrate-binding module (CBM) | 22 | 57 |
| Auxiliary activities (AAs) | 15 | 36 |
| Carbohydrate esterase (CE) | 7 | 18 |
| Polysaccharide lyases (PLs) | 2 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granados-Casas, A.O.; Sastoque, A.P.; Stchigel, A.M.; Fernández-Bravo, A.; Cano-Lira, J.F. Hybrid De Novo Whole-Genome Assembly, Annotation, and Identification of Secondary Metabolite Gene Clusters in the Ex-Type Strain of Chrysosporium keratinophilum. J. Fungi 2023, 9, 389. https://doi.org/10.3390/jof9040389
Granados-Casas AO, Sastoque AP, Stchigel AM, Fernández-Bravo A, Cano-Lira JF. Hybrid De Novo Whole-Genome Assembly, Annotation, and Identification of Secondary Metabolite Gene Clusters in the Ex-Type Strain of Chrysosporium keratinophilum. Journal of Fungi. 2023; 9(4):389. https://doi.org/10.3390/jof9040389
Chicago/Turabian StyleGranados-Casas, Alan Omar, Angie Paola Sastoque, Alberto Miguel Stchigel, Ana Fernández-Bravo, and José Francisco Cano-Lira. 2023. "Hybrid De Novo Whole-Genome Assembly, Annotation, and Identification of Secondary Metabolite Gene Clusters in the Ex-Type Strain of Chrysosporium keratinophilum" Journal of Fungi 9, no. 4: 389. https://doi.org/10.3390/jof9040389