
Global Analyses of Multi-Locus Sequence Typing Data Reveal Geographic Differentiation, Hybridization, and Recombination in the Cryptococcus gattii Species Complex



Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Genotypes and Metadata
2.2. Phylogenetic Distribution of Strains and Genotypes
2.3. Phylogenetic Distribution of Alleles
2.4. Population Genetic Analyses
3. Results
3.1. Geographical and Ecological Distributions
3.2. Geographic AMOVA
3.3. Ecological Niche AMOVA
3.4. DNA Sequence Variation
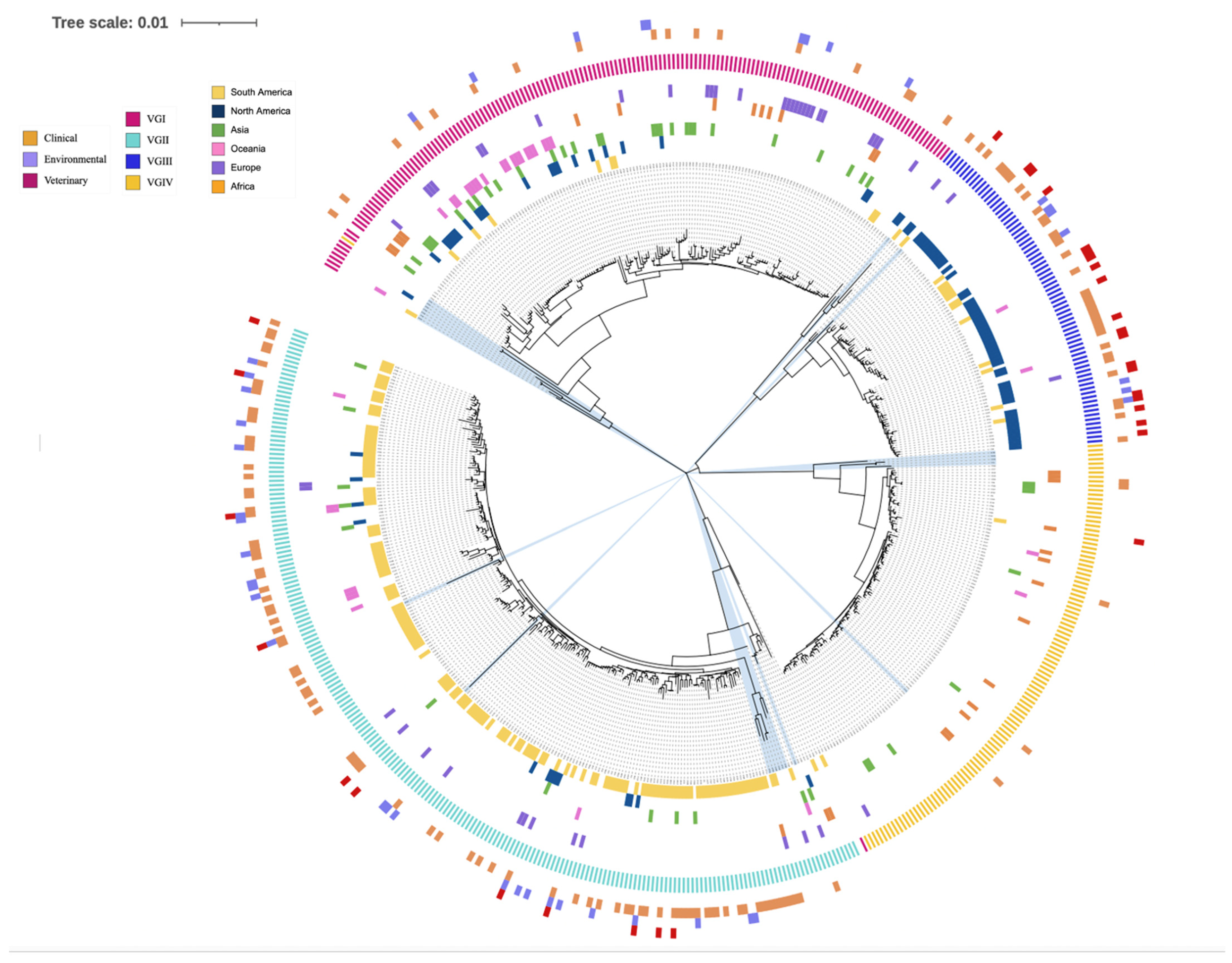
3.5. Phylogenetic Analysis
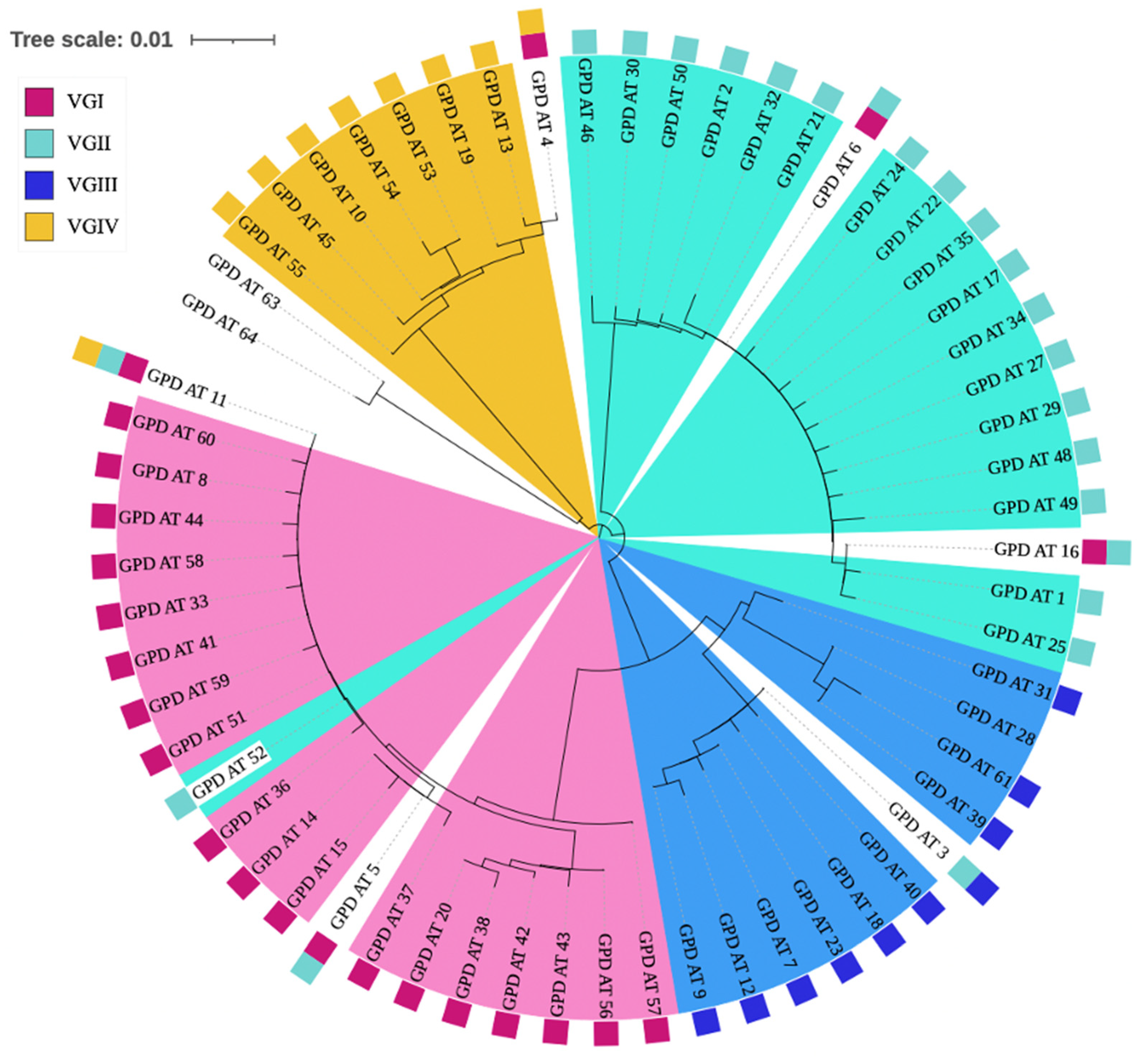
3.6. Inconsistent Allele Type Clustering
3.7. Recombination and Linkage Disequilibrium
4. Discussion
4.1. VG Lineage Distribution
4.2. Geographic and Ecological Structuring
4.3. Hybridization and Recombination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, J.; Lockhart, S.; Chiller, T. Cryptococcus gattii: Where do we go from here? Med. Mycol. 2012, 50, 113–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gushiken, C.A.; Saharia, K.K.; Baddley, W.J. Cryptococcosis. Infect. Dis. Clin. N. Am. 2021, 35, 493–514. [Google Scholar] [CrossRef] [PubMed]
- Hagen, F.; Khayhan, K.; Theelen, B.; Kolecka, A.; Polacheck, I.; Sionov, E.; Falk, R.; Parnmen, S.; Lumbsch, H.T.; Boekhout, T. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 2015, 78, 16–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazera, M.S.; Cavalcanti, M.A.S.; Londero, A.T.; Trilles, L.; Nishikawa, M.M.; Wanke, B. Possible primary ecological niche of Cryptococcus neoformans. Med. Mycol. 2000, 38, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Manosuthi, W.; Banerjee, U.; Zhu, L.-P.; Chen, J.H.; Kohno, S.; Izumikawa, K.; Chen, Y.H.; Sungkanuparph, S.; Harrison, T.S.; et al. Cryptococcosis in Asia. In Cryptococcus: From Human Pathogen to Model Organism; Heitman, J., Kwon-Chung, J., Perfect, J., Casadevall, A., Eds.; ASM Press: Washington, DC, USA, 2011; pp. 287–298. [Google Scholar]
- Randhawa, H.S.; Kowshik, T.; Chowdhary, A.; Preeti Sinha, K.; Khan, Z.U.; Sun, S.; Xu, J. The expanding host tree species spectrum of Cryptococcus gattii and Cryptococcus neoformans and their isolations from surrounding soil in India. Med. Mycol. 2008, 46, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Maziarz, E.K.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. N. Am. 2016, 30, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Kwon-Chung, K.J.; Boekhout, T.; Fell, J.W.; Diaz, M. (1557) Proposal to conserve the name Cryptococcus gattii against C. hondurianus and C. bacillisporus (Basidiomycota, Hymenomycetes, Tremellomycetidae). Taxon 2002, 51, 804–806. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Bennett, J.E. High Prevalence of Cryptococcus neoformans var. gattii in Tropical and Subtropical Regions. Zent. Bakteriol. Mikrobiol. Hygiene. 1. Abt. Originale. A Med. Mikrobiol. Infekt. Parasitol. 1984, 257, 213–218. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J.; Bennett, J.E. Epidemiologic differences between the two varieties of Cryptococcus neoformans. Am. J. Epidemiol. 1984, 120, 123–130. [Google Scholar] [CrossRef]
- Seaton, R.A.; Verma, N.; Naraqi, S.; Wembri, J.P.; Warrell, D.A. Visual loss in immunocompetent patients with Cryptococcus neoformans var. gattii meningitis. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 44–49. [Google Scholar] [CrossRef]
- Speed, B.; Dunt, D. Clinical and Host Differences between Infections with the Two Varieties of Cryptococcus neoformans. Clin. Infect. Dis. 1995, 21, 28–34. [Google Scholar] [CrossRef]
- Ellis, D.H. Cryptococcus neoformans var. gattii in Australia. J. Clin. Microbiol. 1987, 25, 430–431. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.H.; Sorrell, T.C.; Allworth, A.M.; Heath, C.H.; McGregor, A.R.; Papanaoum, K.; Richards, M.J.; Gottlieb, T. Cryptococcal Disease of the CNS in Immunocompetent Hosts: Influence of Cryptococcal Variety on Clinical Manifestations and Outcome. Clin. Infect. Dis. 1995, 20, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Lalloo, D.; Fisher, D.; Naraqi, S.; Laurenson, I.; Temu, P.; Sinha, A.; Saweri, A.; Mavo, B. Cryptococcal meningitis (C. neoformans var. gattii) leading to blindness in previously healthy melanesian adults in Papua New Guinea. QJM Int. J. Med. 1994, 87, 343–349. [Google Scholar] [CrossRef]
- Fyfe, M.; MacDougall, L.; Romney, M.; Starr, M.; Pearce, M.; Mak, S. Cryptococcus gattii infections on Vancouver Island, British Columbia, Canada: Emergence of a tropical fungus in a temperate climate. Can Commun. Dis. Rep. 2008, 34, 1–13. [Google Scholar] [PubMed]
- Stephen, C.; Lester, S.; Black, W.; Fyfe, M.; Raverty, S. Multispecies outbreak of cryptococcosis on southern Vancouver Island, British Columbia. Can. Vet. J. 2002, 43, 792–794. [Google Scholar]
- Galanis, E.; Macdougall, L.; Kidd, S.; Morshed, M. Epidemiology of Cryptococcus gattii, British Columbia, Canada, 1999–2007. Emerg. Infect. Dis. 2010, 16, 251–257. [Google Scholar] [CrossRef]
- Datta, K.; Bartlett, K.H.; Baer, R.; Byrnes, E.; Galanis, E.; Heitman, J.; Hoang, L.; Leslie, M.J.; MacDougall, L.; Magill, S.S.; et al. Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg. Infect. Dis. 2009, 15, 1185–1191. [Google Scholar] [CrossRef]
- Bartlett, K.H.; Kidd, S.E.; Kronstad, J.W. The emergence of Cryptococcus gattii in British Columbia and the Pacific Northwest. Curr. Infect. Dis. Rep. 2008, 10, 58–65. [Google Scholar] [CrossRef]
- Gugnani, H.C.; Mitchell, T.G.; Litvintseva, A.P.; Lengeler, K.B.; Heitman, J.; Kumar, A.; Basu, S.; Paliwal-Joshi, A. Isolation of Cryptococcus gattii and Cryptococcus neoformans var. grubii from the flowers and bark of Eucalyptus trees in India. Med. Mycol. 2005, 43, 565–569. [Google Scholar] [CrossRef] [Green Version]
- Campisi, E.; Mancianti, F.; Pini, G.; Faggi, E.; Gargani, G. Investigation in central Italy of the possible association between Cryptococcus neoformans var. Gattii and Eucalyptus camaldulensis. Eur. J. Epidemiol. 2003, 18, 357–362. [Google Scholar] [CrossRef]
- Kidd, S.E.; Hagen, F.; Tscharke, R.L.; Huynh, M.; Bartlett, K.H.; Fyfe, M.; MacDougall, L.; Boekhout, T.; Kwon-Chung, K.J.; Meyer, W. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc. Natl. Acad. Sci. USA 2004, 101, 17258–17263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukroongreung, S.; Kitiniyom, K.; Nilakul, C.; Tantimavanich, S. Pathogenicity of basidiospores of Filobasidiella neoformans var. neoformans. Med. Mycol. 1998, 36, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hull, C.M.; Heitman, J. Genetics of Cryptococcus neoformans. Annu. Rev. Genet. 2002, 36, 557–615. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.A.; Subaran, R.L.; Nichols, C.B.; Heitman, J. Recapitulation of the Sexual Cycle of the Primary Fungal Pathogen Cryptococcus neoformans var. gattii: Implications for an Outbreak on Vancouver Island, Canada. Eukaryot. Cell 2003, 2, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.A.; Giles, S.S.; Wenink, E.C.; Geunes-Boyer, S.G.; Wright, J.R.; Diezmann, S.; Allen, A.; Stajich, J.E.; Dietrich, F.S.; Perfect, J.R.; et al. Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 2005, 437, 1360–1364. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hull, C.M.; Heitman, J. Sexual reproduction between partners of the same mating type in Cryptococcus neoformans. Nature 2005, 434, 1017–1021. [Google Scholar] [CrossRef]
- Wickes, B.L.; Mayorga, M.E.; Edman, U.; Edman, J.C. Dimorphism and haploid fruiting in Cryptococcus neoformans: Association with the alpha-mating type. Proc. Natl. Acad. Sci. 1996, 93, 7327–7331. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Yan, Z.; Guo, H. Divergence, hybridization, and recombination in the mitochondrial genome of the human pathogenic yeast Cryptococcus gattii. Mol Ecol 2009, 18, 2628–2642. [Google Scholar] [CrossRef]
- Franzot, S.P.; Hamdan, J.S.; Currie, B.P.; Casadevall, A. Molecular epidemiology of Cryptococcus neoformans in Brazil and the United States: Evidence for both local genetic differences and a global clonal population structure. J. Clin. Microbiol. 1997, 35, 2243–2251. [Google Scholar] [CrossRef] [Green Version]
- Kwon-Chung, K.J.; Bennett, J.E. Distribution of α and α mating types of Cryptococcus neoformans among natural and clinical isolates. Am. J. Epidemiol. 1978, 108, 337–340. [Google Scholar] [CrossRef]
- Chowdhary, A.; Hiremath, S.S.; Sun, S.; Kowshik, T.; Randhawa, H.S.; Xu, J. Genetic differentiation, recombination and clonal expansion in environmental populations of Cryptococcus gattii in India. Environ. Microbiol. 2011, 13, 1875–1888. [Google Scholar] [CrossRef] [PubMed]
- Carriconde, F.; Gilgado, F.; Arthur, I.; Ellis, D.; Malik, R.; van de Wiele, N.; Robert, V.; Currie, B.J.; Meyer, W. Clonality and α-a Recombination in the Australian Cryptococcus gattii VGII Population—An Emerging Outbreak in Australia. PLoS ONE 2011, 6, e16936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaocharoen, S.; Ngamskulrungroj, P.; Firacative, C.; Trilles, L.; Piyabongkarn, D.; Banlunara, W.; Poonwan, N.; Chaiprasert, A.; Meyer, W.; Chindamporn, A. Molecular epidemiology reveals genetic diversity amongst isolates of the Cryptococcus neoformans/C. gattii species complex in Thailand. PLoS Negl. Trop. Dis. 2013, 7, e2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, L.T.; Currie, B.J.; Krockenberger, M.; Malik, R.; Meyer, W.; Heitman, J.; Carter, D. Clonality and Recombination in Genetically Differentiated Subgroups of Cryptococcus gattii. Eukaryot Cell 2005, 4, 1403–1409. [Google Scholar] [CrossRef] [Green Version]
- Vélez, N.; Escandón, P. Multilocus sequence typing (MLST) of clinical and environmental isolates of Cryptococcus neoformans and Cryptococcus gattii in six departments of Colombia reveals high genetic diversity. Rev. Soc. Bras. Med. Trop. 2020, 53, e20190422. [Google Scholar] [CrossRef] [PubMed]
- Souto, A.C.; Bonfietti, L.X.; Ferreira-Paim, K.; Trilles, L.; Martins, M.; Ribeiro-Alves, M.; Pham, C.D.; Martins, L.; Dos Santos, W.; Chang, M.; et al. Population Genetic Analysis Reveals a High Genetic Diversity in the Brazilian Cryptococcus gattii VGII Population and Shifts the Global Origin from the Amazon Rainforest to the Semi-arid Desert in the Northeast of Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0004885. [Google Scholar] [CrossRef] [Green Version]
- Meyer, W.; Castañeda, A.; Jackson, S.; Huynh, M.; Castañeda, E. Molecular Typing of IberoAmerican Cryptococcus neoformans Isolates. Emerg. Infect. Dis. 2003, 9, 189–195. [Google Scholar] [CrossRef]
- Boekhout, T.; Theelen, B.; Diaz, M.; Fell, J.W.; Hop, W.C.J.; Abeln, E.C.A.; Dromer, F.; Meyer, W. Hybrid genotypes in the pathogenic yeast Cryptococcus neoformans. Microbiology 2001, 147, 891–907. [Google Scholar] [CrossRef] [Green Version]
- Hong, N.; Chen, M.; Xu, J. Molecular Markers Reveal Epidemiological Patterns and Evolutionary Histories of the Human Pathogenic Cryptococcus. Front. Cell. Infect. Microbiol. 2021, 11, 683670. [Google Scholar] [CrossRef]
- Yamamura, D.; Xu, J. Update on Pulmonary Cryptococcosis. Mycopathologia 2021, 186, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Farrer, R.A.; Chang, M.; Davis, M.J.; Van Dorp, L.; Yang, D.-H.; Shea, T.; Sewell, T.R.; Meyer, W.; Balloux, F.; Edwards, H.M.; et al. A New Lineage of Cryptococcus gattii (VGV) Discovered in the Central Zambezian Miombo Woodlands. mBio 2019, 10, e02306–e02319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroy-Nieto, J.; Bowers, J.R.; Montfort, P.; Adame, G.; Taverna, C.G.; Yaglom, H.; Sykes, J.E.; Brady, S.; Mochon, A.B.; Meyer, W.; et al. Phylogenomic Placement of American Southwest-Associated Clinical and Veterinary Isolates Expands Evidence for Distinct Cryptococcus gattii VGVI. Microorganisms 2022, 10, 1681. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, H.; You, M.; Jenkinson, T.S.; Xu, J.; James, T.Y. Hybridization Facilitates Adaptive Evolution in Two Major Fungal Pathogens. Genes 2020, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Meyer, W.; Aanensen, D.M.; Boekhout, T.; Cogliati, M.; Diaz, M.R.; Esposto, M.C.; Fisher, M.; Gilgado, F.; Hagen, F.; Kaocharoen, S.; et al. Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med. Mycol. 2009, 47, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Meyer, W. International Fungal Multi Locus Sequence Typing Database. Available online: https://mlst.mycologylab.org/page/Home1 (accessed on 15 January 2023).
- Hitchcock, M.; Xu, J. Analyses of the Global Multilocus Genotypes of the Human Pathogenic Yeast Cryptococcus neoformans Species Complex. Genes 2022, 13, 2045. [Google Scholar] [CrossRef]
- Xess, I.; Pandey, M.; Dabas, Y.; Agarwal, R.; Das, S.; Srivastava, P.M.V.; Thakur, R.; Sharma, S.; Mani, P.; Biswas, A.; et al. Multilocus Sequence Typing of Clinical Isolates of Cryptococcus from India. Mycopathologia 2021, 186, 199–211. [Google Scholar] [CrossRef]
- Rocha, D.F.S.; Cruz, K.S.; Santos, C.; Menescal, L.S.F.; Neto, J.; Pinheiro, S.B.; Silva, L.M.; Trilles, L.; Braga de Souza, J.V. MLST reveals a clonal population structure for Cryptococcus neoformans molecular type VNI isolates from clinical sources in Amazonas, Northern-Brazil. PLoS ONE 2018, 13, e0197841. [Google Scholar] [CrossRef] [Green Version]
- Moslem, M.; Fatahinia, M.; Kiasat, N.; Mahmoudabadi, A.Z. Genotypic diversity of Iranian Cryptococcus neoformans using multilocus sequence typing (MLST) and susceptibility to antifungals. Mol. Biol. Rep. 2021, 48, 4201–4208. [Google Scholar] [CrossRef]
- Huang, C.; Tsui, C.K.M.; Chen, M.; Pan, K.; Li, X.; Wang, L.; Chen, M.; Zheng, Y.; Zheng, D.; Chen, X.; et al. Emerging Cryptococcus gattii species complex infections in Guangxi, southern China. PLoS Negl. Trop. Dis. 2020, 14, e0008493. [Google Scholar] [CrossRef]
- Firacative, C.; Roe, C.C.; Malik, R.; Ferreira-Paim, K.; Escandón, P.; Sykes, J.E.; Castañón-Olivares, L.R.; Contreras-Peres, C.; Samayoa, B.; Sorrell, T.C.; et al. MLST and Whole-Genome-Based Population Analysis of Cryptococcus gattii VGIII Links Clinical, Veterinary and Environmental Strains, and Reveals Divergent Serotype Specific Sub-populations and Distant Ancestors. PLoS Negl. Trop. Dis. 2016, 10, e0004861. [Google Scholar] [CrossRef] [Green Version]
- Bellet, V.; Roger, F.; Krasteva, D.; Gouveia, T.; Drakulovski, P.; Pottier, C.; Bertout, S. Multilocus sequence typing of strains from the Cryptococcus gattii species complex from different continents. Mycoses 2022, 65, 88–96. [Google Scholar] [CrossRef]
- Meyer, W.; Trilles, L. Genotyping of the Cryptococcus neoformans/Cryptococcus gattii species complex. Aust. Biochem. 2010, 41, 12–16. [Google Scholar]
- Wu, S.-Y.; Kang, M.; Liu, Y.; Chen, Z.-X.; Xiao, Y.-L.; He, C.; Ma, Y. Molecular epidemiology and antifungal susceptibilities of Cryptococcus species isolates from HIV and non-HIV patients in Southwest China. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Cao, J.R.; Xue, X.Y.; Wu, H.; Wang, L.F.; Guo, L.; Shen, D.X. Clinical and microbiological characteristics of Cryptococcus gattii isolated from 7 hospitals in China. BMC Microbiol. 2020, 20, 73. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, F.H.; de Paula, D.A.J.; Menezes, I.D.G.; Favalessa, O.C.; Hahn, R.C.; de Almeida, A.D.B.P.F.; Sousa, V.R.F.; Nakazato, L.; Dutra, V. Genetic Diversity of the Cryptococcus gattii Species Complex in Mato Grosso State, Brazil. Mycopathologia 2019, 184, 45–51. [Google Scholar] [CrossRef]
- Firacative, C.; Torres, G.; Meyer, W.; Escandón, P. Clonal Dispersal of Cryptococcus gattii VGII in an Endemic Region of Cryptococcosis in Colombia. J. Fungi 2019, 5, 32. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.T.; Xu, Y.C.; Wang, H.Z.; Li, T.S. Molecular epidemiology of Cryptococcus neoformans and Cryptococcus gattii in China between 2007 and 2013 using multilocus sequence typing and the DiversiLab system. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 753–762. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Lei, Y.; Kang, M.; Xiao, Y.-L.; Chen, Z.-X. Molecular characterisation of clinical Cryptococcus neoformans and Cryptococcus gattii isolates from Sichuan province, China. Mycoses 2015, 58, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Bellet, V.; Kassi, F.K.; Krasteva, D.; Roger, F.; Drakulovski, P.; Mossou, C.; Kouakou, G.A.; Doumbia, A.; Delaporte, E.; Menan, H.; et al. First report of cryptococcosis due to Cryptococcus gattii sensu stricto VGI in an Ivorian HIV negative patient. J. Med. Mycol. 2021, 31, 101113. [Google Scholar] [CrossRef]
- Lin, K.-H.; Lin, Y.-P.; Ho, M.-W.; Chen, Y.-C.; Chung, W.-H. Molecular epidemiology and phylogenetic analyses of environmental and clinical isolates of Cryptococcus gattii sensu lato in Taiwan. Mycoses 2021, 64, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Xiao, M.; Chen, S.; Kong, F.; Dou, H.-T.; Wang, H.; Xiao, Y.-L.; Kang, M.; Sun, Z.-Y.; Hu, Z.-D.; et al. Predominance of Cryptococcus neoformans var. grubii multilocus sequence type 5 and emergence of isolates with non-wild-type minimum inhibitory concentrations to fluconazole: A multi-centre study in China. Clin. Microbiol. Infect. 2016, 22, 887.e1–887.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos, V.A.; Martins, L.M.S.; Fontes, A.C.L.; Reuwsaat, J.C.V.; Squizani, E.D.; de Sousa Araújo, G.R.; Frases, S.; Staats, C.C.; Schrank, A.; Kmetzsch, L.; et al. Genotypic and Phenotypic Diversity of Cryptococcus gattii VGII Clinical Isolates and Its Impact on Virulence. Front. Microbiol. 2018, 9, 132. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, M.; Desnos-Ollivier, M.; McCormick-Smith, I.; Rickerts, V.; Ferreira-Paim, K.; Meyer, W.; Boekhout, T.; Hagen, F.; Theelen, B.; Inácio, J.; et al. Genotypes and population genetics of Cryptococcus neoformans and Cryptococcus gattii species complexes in Europe and the Mediterranean area. Fungal Genet. Biol. 2019, 129, 16–29. [Google Scholar] [CrossRef]
- Pinheiro, S.B.; Sousa, E.S.; Cortez, A.C.A.; da Silva Rocha, D.F.; Menescal, L.S.F.; Chagas, V.S.; Gómez, A.S.P.; Cruz, K.S.; Santos, L.O.; Alves, M.J.; et al. Cryptococcal meningitis in non-HIV patients in the State of Amazonas, Northern Brazil. Braz. J. Microbiol. 2021, 52, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Brito-Santos, F.; Reis, R.S.; Coelho, R.A.; Almeida-Paes, R.; Pereira, S.A.; Trilles, L.; Meyer, W.; Wanke, B.; Lazéra, M.D.S.; Gremião, I.D.F. Cryptococcosis due to Cryptococcus gattii VGII in southeast Brazil: The One Health approach revealing a possible role for domestic cats. Med. Mycol. Case Rep. 2019, 24, 61–64. [Google Scholar] [CrossRef]
- Brito-Santos, F.; Barbosa, G.G.; Trilles, L.; Nishikawa, M.M.; Wanke, B.; Meyer, W.; Carvalho-Costa, F.A.; Lazéra Mdos, S. Environmental isolation of Cryptococcus gattii VGII from indoor dust from typical wooden houses in the deep Amazonas of the Rio Negro basin. PLoS ONE 2015, 10, e0115866. [Google Scholar] [CrossRef] [Green Version]
- Lizarazo, J.; Escandón, P.; Agudelo, C.I.; Firacative, C.; Meyer, W.; Castañeda, E. Retrospective study of the epidemiology and clinical manifestations of Cryptococcus gattii infections in Colombia from 1997–2011. PLoS Negl. Trop. Dis. 2014, 8, e3272. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.-f.; Zhang, P.-p.; Wang, J.; Yang, Q.; Qu, T.-t. Clinical and microbiological characteristics of cryptococcosis at an university hospital in China from 2013 to 2017. Braz. J. Infect. Dis. 2020, 24, 7–12. [Google Scholar] [CrossRef]
- Brito-Santos, F.; Trilles, L.; Firacative, C.; Wanke, B.; Carvalho-Costa, F.A.; Nishikawa, M.M.; Campos, J.P.; Junqueira, A.C.V.; Souza, A.C.; Lazéra, M.D.S.; et al. Indoor Dust as a Source of Virulent Strains of the Agents of Cryptococcosis in the Rio Negro Micro-Region of the Brazilian Amazon. Microorganisms 2020, 8, 682. [Google Scholar] [CrossRef]
- Fu, Y.; Xu, M.; Zhou, H.; Yao, Y.; Zhou, J.; Pan, Z. Microbiological and clinical characteristics of cryptococcemia: A retrospective analysis of 85 cases in a Chinese hospital. Med. Mycol. 2019, 58, 478–484. [Google Scholar] [CrossRef]
- Kassi, F.K.; Drakulovski, P.; Bellet, V.; Roger, F.; Chabrol, A.; Krasteva, D.; Doumbia, A.; Landman, R.; Kakou, A.; Reynes, J.; et al. Cryptococcus genetic diversity and mixed infections in Ivorian HIV patients: A follow up study. PLOS Negl. Trop. Dis. 2019, 13, e0007812. [Google Scholar] [CrossRef] [Green Version]
- Schmertmann, L.J.; Danesi, P.; Monroy-Nieto, J.; Bowers, J.; Engelthaler, D.M.; Malik, R.; Meyer, W.; Krockenberger, M.B. Jet-Setting Koalas Spread Cryptococcus gattii VGII in Australia. mSphere 2019, 4, e00216-19. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agapow, P.-M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Lockhart, S.R.; Iqbal, N.; Harris, J.R.; Grossman, N.T.; DeBess, E.; Wohrle, R.; Marsden-Haug, N.; Vugia, D.J. Cryptococcus gattii in the United States: Genotypic Diversity of Human and Veterinary Isolates. PLoS ONE 2013, 8, e74737. [Google Scholar] [CrossRef]
- Cogliati, M. Global Molecular Epidemiology of Cryptococcus neoformans and Cryptococcus gattii: An Atlas of the Molecular Types. Science 2013, 2013, 675213. [Google Scholar] [CrossRef] [Green Version]
- Litvintseva, A.P.; Thakur, R.; Reller, L.B.; Mitchell, T.G. Prevalence of Clinical Isolates of Cryptococcus gattii Serotype C among Patients with AIDS in Sub-Saharan Africa. J. Infect. Dis. 2005, 192, 888–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J. Fundamentals of fungal molecular population genetic analyses. Curr. Issues Mol. Biol. 2006, 8, 75–89. [Google Scholar] [PubMed]
- Fisher, M.C.; Alastruey-Izquierdo, A.; Berman, J.; Bicanic, T.; Bignell, E.M.; Bowyer, P.; Bromley, M.; Brüggemann, R.; Garber, G.; Cornely, O.A.; et al. Tackling the emerging threat of antifungal resistance to human health. Nat. Rev. Microbiol. 2022, 20, 557–571. [Google Scholar] [CrossRef]
- Xu, J. Assessing global fungal threats to humans. mLife 2022, 1, 223–240. [Google Scholar] [CrossRef]
- Kidd, S.E.; Bach, P.J.; Hingston, A.O.; Mak, S.; Chow, Y.; MacDougall, L.; Kronstad, J.W.; Bartlett, K.H. Cryptococcus gattii Dispersal Mechanisms, British Columbia, Canada. Emerg. Infect. Dis. 2007, 13, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velegraki, A.; Kiosses, V.G.; Kansouzidou, A.; Smilakou, S.; Mitroussia-Ziouva, A.; Legakis, N.J. Prospective use of RFLP analysis on amplified Cryptococcus neoformans URA5 gene sequences for rapid identification of varieties and serotypes in clinical samples. Med. Mycol. 2001, 39, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Farrer, R.A.; Desjardins, C.A.; Sakthikumar, S.; Gujja, S.; Saif, S.; Zeng, Q.; Chen, Y.; Voelz, K.; Heitman, J.; May, R.C.; et al. Genome Evolution and Innovation across the Four Major Lineages of Cryptococcus gattii. mBio 2015, 6, e00868-15. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.C.; Hanage, W.P.; de Hoog, S.; Johnson, E.; Smith, M.D.; White, N.J.; Vanittanakom, N. Low Effective Dispersal of Asexual Genotypes in Heterogeneous Landscapes by the Endemic Pathogen Penicillium marneffei. PLOS Pathog. 2005, 1, e20. [Google Scholar] [CrossRef] [Green Version]
- Goddard, M.R.; Godfray, H.C.; Burt, A. Sex increases the efficacy of natural selection in experimental yeast populations. Nature 2005, 434, 636–640. [Google Scholar] [CrossRef]
- Billmyre, R.B.; Croll, D.; Li, W.; Mieczkowski, P.; Carter, D.A.; Cuomo, C.A.; Kronstad, J.W.; Heitman, J. Highly Recombinant VGII Cryptococcus gattii Population Develops Clonal Outbreak Clusters through both Sexual Macroevolution and Asexual Microevolution. mBio 2014, 5, e01494-14. [Google Scholar] [CrossRef] [Green Version]
- Hiremath, S.S.; Chowdhary, A.; Kowshik, T.; Randhawa, H.S.; Sun, S.; Xu, J. Long-distance dispersal and recombination in environmental populations of Cryptococcus neoformans var. grubii from India. Microbiology 2008, 154 Pt 5, 1513–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, F.; Ceresini, P.C.; Polacheck, I.; Ma, H.; van Nieuwerburgh, F.; Gabaldón, T.; Kagan, S.; Pursall, E.R.; Hoogveld, H.L.; van Iersel, L.J.J.; et al. Ancient Dispersal of the Human Fungal Pathogen Cryptococcus gattii from the Amazon Rainforest. PLoS ONE 2013, 8, e71148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon-Chung, K.J. A New Genus, Filobasidiella, The Perfect State of Cryptococcus neoformans. Mycologia 1975, 67, 1197–1200. [Google Scholar] [CrossRef]
- Kwon-Chung, K.J. A New Species of Filobasidiella, The Sexual State of Cryptococcus neoformans B and C Serotypes. Mycologia 1976, 68, 942–946. [Google Scholar] [CrossRef]
- Halliday, C.L.; Bui, T.; Krockenberger, M.; Malik, R.; Ellis, D.H.; Carter, D.A. Presence of α and Mating Types in Environmental and Clinical Collections of Cryptococcus neoformans var. gattii Strains from Australia. J. Clin. Microbiol. 1999, 37, 2920–2926. [Google Scholar] [CrossRef] [Green Version]
- You, M.; Xu, J. What Are the Best Parents for Hybrid Progeny? An Investigation into the Human Pathogenic Fungus Cryptococcus. J. Fungi 2021, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Lin, Y.M.; Dobrin, A.; Xu, J. Genetic and Phenotypic Diversities in Experimental Populations of Diploid Inter-Lineage Hybrids in the Human Pathogenic Cryptococcus. Microorganisms 2021, 9, 1579. [Google Scholar] [CrossRef]
- Samarasinghe, H.; Xu, J. Hybrids and hybridization in the Cryptococcus neoformans and Cryptococcus gattii species complexes. Infect. Genet. Evol. 2018, 66, 245–255. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sample Group | No. of Recorded ST | No. of ST with Geographical Data | No. of Isolates with Geographical Data | Found in No. of Continents | Found in No. of Countries | No. of ST with Ecological Data | No. of Isolates with Ecological Data | Found in No. of Ecological Sources |
|---|---|---|---|---|---|---|---|---|
| Total CGSC | 566 | 375 | 1202 | 6 | 45 | 188 | 788 | 3 |
| VGI | 160 | 97 | 225 | 6 | 31 | 23 | 103 | 2 |
| VGII | 218 | 181 | 721 | 6 | 25 | 105 | 530 | 3 |
| VGIII | 75 | 72 | 221 | 4 | 12 | 56 | 144 | 3 |
| VGIV | 111 | 24 | 34 | 5 | 9 | 7 | 10 | 2 |
| ST previously unassigned to a lineage | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Region | n | % | VGI | VGII | VGIII | VGIV | Region | n | % | VGI | VGII | VGIII | VGIV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Africa | 86 | 7.15% | 20 | 45 | 0 | 21 | North America | 179 | 14.89% | 25 | 15 | 139 | 0 |
| Ivory Coast | 45 | 3.74% | 2 | 43 | 0 | 0 | USA | 141 | 11.73% | 20 | 7 | 114 | 0 |
| DRC | 12 | 1.00% | 10 | 2 | 0 | 0 | Mexico | 25 | 2.08% | 1 | 0 | 24 | 0 |
| South Africa | 12 | 1.00% | 4 | 0 | 0 | 8 | Canada | 10 | 0.83% | 3 | 7 | 0 | 0 |
| Zimbabwe | 12 | 1.00% | 0 | 0 | 0 | 12 | Aruba | 1 | 0.08% | 0 | 1 | 0 | 0 |
| Rwanda | 3 | 0.25% | 2 | 0 | 0 | 1 | Cuba | 1 | 0.08% | 1 | 0 | 0 | 0 |
| Kenya | 2 | 0.17% | 2 | 0 | 0 | 0 | Guatemala | 1 | 0.08% | 0 | 0 | 1 | 0 |
| Asia | 196 | 16.31% | 87 | 141 | 0 | 9 | Oceania | 159 | 13.23% | 24 | 129 | 4 | 2 |
| Taiwan | 111 | 9.23% | 44 | 66 | 0 | 1 | Australia | 153 | 12.73% | 20 | 129 | 2 | 2 |
| China | 43 | 3.58% | 28 | 14 | 0 | 0 | New Zealand | 3 | 0.25% | 1 | 0 | 2 | 0 |
| Thailand | 17 | 1.41% | 3 | 14 | 0 | 0 | Papa New Guinea | 3 | 0.25% | 3 | 0 | 0 | 0 |
| India | 10 | 0.83% | 6 | 0 | 0 | 4 | |||||||
| Malaysia | 10 | 0.83% | 3 | 3 | 0 | 4 | |||||||
| Singapore | 3 | 0.25% | 3 | 0 | 0 | 0 | |||||||
| Japan | 1 | 0.08% | 0 | 1 | 0 | 0 | |||||||
| South Korea | 1 | 0.08% | 0 | 1 | 0 | 0 | |||||||
| Europe | 65 | 5.41% | 44 | 17 | 3 | 1 | South America | 517 | 43.01% | 25 | 416 | 75 | 1 |
| Spain | 14 | 1.16% | 12 | 1 | 1 | 0 | Brazil | 349 | 29.03% | 9 | 340 | 0 | 0 |
| Greece | 11 | 0.92% | 8 | 3 | 0 | 0 | Colombia | 145 | 12.06% | 13 | 63 | 68 | 1 |
| France | 9 | 0.75% | 5 | 4 | 0 | 0 | Argentina | 10 | 0.83% | 1 | 7 | 2 | 0 |
| Italy | 9 | 0.75% | 9 | 0 | 0 | 0 | Venezuela | 5 | 0.42% | 0 | 3 | 2 | 0 |
| The Netherlands | 8 | 0.67% | 2 | 6 | 0 | 0 | Paraguay | 2 | 0.17% | 0 | 0 | 2 | 0 |
| Germany | 5 | 0.42% | 2 | 1 | 2 | 0 | Peru | 2 | 0.17% | 2 | 0 | 0 | 0 |
| Portugal | 3 | 0.25% | 3 | 0 | 0 | 0 | Uruguay | 2 | 0.17% | 0 | 2 | 0 | 0 |
| Belgium | 2 | 0.17% | 2 | 0 | 0 | 0 | Chile | 1 | 0.08% | 0 | 0 | 1 | 0 |
| Switzerland | 2 | 0.17% | 1 | 1 | 0 | 0 | French Guiana | 1 | 0.08% | 0 | 1 | 0 | 0 |
| Denmark | 1 | 0.08% | 0 | 1 | 0 | 0 | |||||||
| Sweden | 1 | 0.08% | 0 | 0 | 0 | 1 |
| Distribution Patterns | Specific Continent(s)/Ecological Niche(s) | Number of Sequence Types | Number of Isolates |
|---|---|---|---|
| Geographic | |||
| In all six continents | Africa + Asia + Europe + N America + S America + Oceania | 0 | 0 |
| In five continents only | 0 | 0 | |
| In four continents only | |||
| Asia + Europe + N America + S America | 2 | 50 | |
| Asia + Europe + N America + Oceania | 1 | 11 | |
| Other combinations of four continents | 0 | 0 | |
| In three continents only | |||
| Africa + Asia + Europe | 3 | 56 | |
| Asia + Europe + S America | 1 | 8 | |
| Asia + N America + S America | 2 | 186 | |
| Asia + N America + Oceania | 1 | 4 | |
| Other combinations of three continents | 0 | 0 | |
| In two continents only | |||
| Africa + Asia | 1 | 2 | |
| Africa + Europe | 1 | 3 | |
| Africa + S America | 1 | 3 | |
| Asia + Europe | 2 | 5 | |
| Asia + N America | 3 | 41 | |
| Asia + S America | 3 | 42 | |
| Asia + Oceania | 1 | 4 | |
| Europe + N America | 2 | 4 | |
| Europe + S America | 2 | 8 | |
| N America + S America | 5 | 95 | |
| N America + Oceania | 2 | 6 | |
| S America + Oceania | 1 | 16 | |
| Other combinations of two continents | 0 | 0 | |
| In one continent only | |||
| Africa | 24 | 32 | |
| Asia | 30 | 74 | |
| Europe | 31 | 45 | |
| N America | 75 | 131 | |
| S America | 156 | 331 | |
| Oceania | 25 | 45 | |
| Ecological niches | |||
| In all three niches | 5 | 237 | |
| In two niches only | |||
| Clinical + Veterinary | 5 | 31 | |
| Clinical + Environmental | 10 | 165 | |
| Veterinary + Environmental | 2 | 9 | |
| In one niche only | |||
| Clinical | 123 | 255 | |
| Veterinary | 19 | 22 | |
| Environmental | 24 | 69 |
| Non-Clone-Corrected | Clone-Corrected | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| df | MS | Est.Var | % | p-Value | df | MS | Est.Var | % | p-Value | |
| Total CGSC | ||||||||||
| Among continents | 5 | 98.373 | 0.540 | 17% | 0.001 | 5 | 14.283 | 0.175 | 5% | 0.001 |
| Within continents | 1196 | 2.646 | 2.649 | 83% | 0.001 | 417 | 3.100 | 3.100 | 95% | 0.001 |
| Total | 1201 | 3.045 | 3.189 | 422 | 3.23 | 3.275 | ||||
| VGI | ||||||||||
| Among continents | 5 | 20.456 | 0.532 | 21% | 0.001 | 5 | 10.329 | 0.408 | 15% | 0.001 |
| Within continents | 219 | 2.050 | 2.050 | 79% | 0.001 | 115 | 2.264 | 2.264 | 85% | 0.001 |
| Total | 224 | 2.46 | 2.582 | 120 | 2.6 | 2.672 | ||||
| VGII | ||||||||||
| Among continents | 4 | 46.516 | 0.417 | 16% | 0.001 | 4 | 4.798 | 0.102 | 4% | 0.001 |
| Within continents | 713 | 2.178 | 2.178 | 84% | 0.001 | 190 | 2.723 | 2.723 | 96% | 0.001 |
| Total | 717 | 2.42 | 2.594 | 194 | 2.766 | 2.826 | ||||
| VGIII | ||||||||||
| Among continents | 1 | 33.565 | 0.321 | 12% | 0.001 | 1 | 4.613 | 0.082 | 3% | 0.066 |
| Within continents | 212 | 2.318 | 2.318 | 88% | 0.001 | 71 | 2.657 | 2.657 | 97% | 0.066 |
| Total | 213 | 2.46 | 2.639 | 72 | 2.68 | 2.739 | ||||
| VGIV | ||||||||||
| Among continents | 1 | 4.217 | 0.173 | 8% | 0.022 | 1 | 2.772 | 0.057 | 3% | 0.196 |
| Within continents | 28 | 2.036 | 2.036 | 92% | 0.022 | 19 | 2.210 | 2.210 | 97% | |
| Total | 29 | 2.11 | 2.209 | 20 | 2.2 | 2.267 | ||||
| Non-Clone-Corrected | Clone-Corrected | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total CGSC | ||||||||||
| Africa | Asia | Europe | Oceania | N. America | Africa | Asia | Europe | Oceania | N. America | |
| Asia | 0.119 *** | 0.093 *** | ||||||||
| Europe | 0.322 *** | 0.220 *** | 0.184 *** | 0.043 *** | ||||||
| Oceania | 0.410 *** | 0.362 *** | 0.183 *** | 0.053 *** | 0.035 *** | 0.056 *** | ||||
| N. America | 0.307 *** | 0.255 *** | 0.110 *** | 0.061 *** | 0.120 *** | 0.015 *** | 0.070 *** | 0.075 *** | ||
| S. America | 0.223 *** | 0.169 *** | 0.053 *** | 0.128 *** | 0.065 *** | 0.036 *** | 0.026 *** | 0.101 *** | 0.034 *** | 0.034 *** |
| VGI | ||||||||||
| Asia | 0.173 *** | 0.051 *** | ||||||||
| Europe | 0.095 *** | 0.210 *** | 0.083 *** | 0.062 *** | ||||||
| Oceania | 0.453 *** | 0.307 *** | 0.491 *** | 0.403 *** | 0.220 *** | 0.412 *** | ||||
| N. America | 0.189 *** | 0.104 *** | 0.242 *** | 0.130 *** | 0.153 *** | 0.037 *** | 0.165 *** | 0.123 *** | ||
| S. America | 0.145 *** | 0.093 *** | 0.161 *** | 0.237 *** | 0.045 *** | 0.089 *** | 0.010 *** | 0.102 *** | 0.262 *** | 0.045 *** |
| VGII | ||||||||||
| Asia | N/A | N/A | ||||||||
| Europe | N/A | 0.191 *** | N/A | 0.009 *** | ||||||
| Oceania | N/A | 0.320 *** | 0.388 *** | N/A | 0.015 *** | 0.061 *** | ||||
| N. America | N/A | 0.229 *** | 0.078 *** | 0.168 *** | N/A | 0.027 *** | 0.020 *** | 0.066 *** | ||
| S. America | N/A | 0.135 *** | 0.051 *** | 0.158 *** | 0.025 *** | N/A | 0.00 *** | 0.049 *** | 0.047 *** | 0.051 *** |
| VGIII | ||||||||||
| Asia | N/A | N/A | ||||||||
| Europe | N/A | N/A | N/A | N/A | ||||||
| Oceania | N/A | N/A | N/A | N/A | N/A | N/A | ||||
| N. America | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | ||
| S. America | N/A | N/A | N/A | N/A | 0.122 *** | N/A | N/A | N/A | N/A | 0.030 |
| VGIV | ||||||||||
| Asia | 0.078 * | 0.025 | ||||||||
| Europe | N/A | N/A | N/A | N/A | ||||||
| Oceania | N/A | N/A | N/A | N/A | N/A | N/A | ||||
| N. America | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | ||
| S. America | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Non-Clone-Corrected | Clone-Corrected | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| df | MS | Est.Var | % | p-Value | df | MS | Est.Var | % | p-Value | |
| Australia | ||||||||||
| Among ecological niches | 2 | 23.8 | 0.32 | 22% | 0.001 | N/A | N/A | N/A | N/A | N/A |
| Within ecological niches | 214 | 1.1 | 1.14 | 78% | 0.001 | N/A | N/A | N/A | N/A | N/A |
| Total | 216 | 1.36 | 1.46 | N/A | N/A | N/A | ||||
| Brazil | ||||||||||
| Among ecological niches | 2 | 32.1 | 0.24 | 11% | 0.001 | 2 | 2.99 | 0.02 | 1% | 0.318 |
| Within ecological niches | 400 | 1.99 | 1.99 | 89% | 0.001 | 97 | 2.66 | 2.66 | 99% | 0.318 |
| Total | 402 | 2.12 | 2.24 | 99 | 2.67 | 2.67 | ||||
| Colombia | ||||||||||
| Among ecological niches | 2 | 28.5 | 0.24 | 10% | 0.001 | 1 | 2.73 | 0.0 | 0% | 0.502 |
| Within ecological niches | 332 | 2.2 | 2.16 | 90% | 0.001 | 29 | 3.03 | 3.03 | 100% | 0.502 |
| Total | 334 | 3 | 2.31 | 30 | 2.40 | 3.03 | ||||
| Taiwan | ||||||||||
| Among ecological niches | 2 | 19.2 | 0.24 | 13% | 0.001 | N/A | N/A | N/A | N/A | N/A |
| Within ecological niches | 255 | 1.58 | 1.58 | 87% | 0.001 | N/A | N/A | N/A | N/A | N/A |
| Total | 257 | 1.82 | N/A | N/A | N/A | |||||
| USA | ||||||||||
| Among ecological niches | 2 | 29.6 | 0.26 | 10% | 0.001 | 1 | 2.44 | 0.0 | 0% | 0.54 |
| Within ecological niches | 320 | 2.29 | 2.29 | 90% | 0.001 | 45 | 2.78 | 2.78 | 100% | 0.54 |
| Total | 322 | 2.46 | 2.56 | 46 | 2.78 | 2.78 | ||||
| Gene | Gene Name | Chromosome | Length (bp) | Total Allele Number in CGSC | Number of Shared Alleles between VG Lineages |
|---|---|---|---|---|---|
| CAP59 | Capsular-associated protein | 1 | 556–558 | 95 | 1 |
| GPD1 | Glyceraldehyde-3-phosphate dehydrogenase | 7 | 544–549 | 61 | 6 |
| IGS1 | Ribosomal RNA intergenic spacer | 2 | 624–706 | 110 | 4 |
| LAC1 | Laccase | 8 | 472–475 | 63 | 2 |
| PLB1 | Phospholipase | 12 | 532–535 | 46 | 1 |
| SOD1 | Cu, Zn superoxide dismutase | 5 | 700–713 | 136 | 5 |
| URA5 | Orotidine monophosphate pyrophosphorylase | 8 | 637–639 | 59 | 1 |
| ST | No. of Inconsistent ATs | No. of VG Lineages Represented | Originally Assigned VG Lineage | CAP59 | GPD1 | IGS1 | LAC1 | PLB1 | SOD1 | URA5 |
|---|---|---|---|---|---|---|---|---|---|---|
| ST11 | 1 | 2 | VGIV | 17 | 10 | 2 | 1 | 3 | 93 | 11 |
| ST53 | 1 | 2 | VGI | 16 | 5 | 40 | 13 | 5 | 32 | 12 |
| ST167 | 1 | 2 | VGII | 2 | 3 | 31 | 4 | 1 | 8 | 7 |
| ST181 | 1 | 2 | VGII | 8 | 3 | 22 | 4 | 2 | 87 | 3 |
| ST223 | 1 | 2 | VGIII | 42 | 28 | 61 | 41 | 31 | 51 | 17 |
| ST335 | 1 | 2 | VGIII | 29 | 12 | 11 | 9 | 13 | 28 | 22 |
| ST350 | 1 | 2 | VGII | 5 | 11 | 25 | 4 | 16 | 16 | 6 |
| ST351 | 4 | 2 | VGI | 44 | 16 | 59 | 4 | 13 | 14 | 14 |
| ST355 | 1 | 2 | VGII | 3 | 5 | 57 | 4 | 18 | 58 | 2 |
| ST356 | 1 | 2 | VGI | 16 | 6 | 3 | 5 | 5 | 45 | 12 |
| ST357 | 1 | 2 | VGII | 1 | 6 | 25 | 4 | 2 | 45 | 7 |
| ST386 | 1 | 2 | VGIV | 63 | 11 | 2 | 1 | 3 | 62 | 11 |
| ST400 | 1 | 2 | VGII | 2 | 52 | 56 | 21 | 1 | 90 | 7 |
| ST402 | 6 | 3 | VGIV | 25 | 11 | 46 | 13 | 13 | 73 | 11 |
| ST408 | 1 | 2 | VGI | 71 | 42 | 90 | 6 | 5 | 113 | 38 |
| ST409 | 1 | 2 | VGII | 2 | 52 | 56 | 21 | 1 | 8 | 7 |
| ST419 | 1 | 2 | VGIV | 23 | 4 | 2 | 17 | 7 | 35 | 34 |
| ST431 | 2 | 3 | VGI | 44 | 4 | 59 | 13 | 13 | 68 | 15 |
| ST451 | 1 | 2 | VGII | 8 | 6 | 14 | 4 | 2 | 6 | 3 |
| ST508 | 1 | 2 | VGIV | 23 | 4 | 103 | 17 | 44 | 35 | 34 |
| ST524 | 2 | 3 | VGI | 44 | 4 | 102 | 13 | 13 | 68 | 15 |
| ST528 | 1 | 2 | VGI | 71 | 56 | 90 | 6 | 5 | 113 | 38 |
| ST550 | 1 | 2 | VGI | 16 | 5 | 12 | 5 | 5 | 32 | 3 |
| Population | Number | Phylogenetic Compatibility (% of 21 Pairs) | Index of Association |
|---|---|---|---|
| Global CGSC | 566 | 0 | 0.82 *** |
| Global VGI | 160 | 0 | 1.27 *** |
| Global VGII | 218 | 0 | 0.28 *** |
| Global VGIII | 75 | 9.5% | 1.46 *** |
| Global VGIV | 111 | 23.8% | 0.14 ** |
| Africa CGSC | 31 | 52.4% | 2.10 *** |
| Asia CGSC | 50 | 71.4% | 1.99 *** |
| Europe CGSC | 46 | 76.1% | 2.40 *** |
| Oceania CGSC | 34 | 90.4% | 3.10 *** |
| North America CGSC | 94 | 14.2% | 1.96 *** |
| South America CGSC | 174 | 0 | 0.55 *** |
| Asia VGI | 29 | 76.1% | 1.63 *** |
| Europe VGI | 28 | 85.7% | 1.51 *** |
| Oceania VGI | 21 | 90.4% | 1.05 *** |
| North America VGI | 23 | 90.4% | 1.43 *** |
| North America VGII | 59 | 19% | 1.37 *** |
| South America VGII | 148 | 0 | 1.60 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hitchcock, M.; Xu, J. Global Analyses of Multi-Locus Sequence Typing Data Reveal Geographic Differentiation, Hybridization, and Recombination in the Cryptococcus gattii Species Complex. J. Fungi 2023, 9, 276. https://doi.org/10.3390/jof9020276
Hitchcock M, Xu J. Global Analyses of Multi-Locus Sequence Typing Data Reveal Geographic Differentiation, Hybridization, and Recombination in the Cryptococcus gattii Species Complex. Journal of Fungi. 2023; 9(2):276. https://doi.org/10.3390/jof9020276
Chicago/Turabian StyleHitchcock, Megan, and Jianping Xu. 2023. "Global Analyses of Multi-Locus Sequence Typing Data Reveal Geographic Differentiation, Hybridization, and Recombination in the Cryptococcus gattii Species Complex" Journal of Fungi 9, no. 2: 276. https://doi.org/10.3390/jof9020276