
Exploring the Molecular Underpinnings of Cancer-Causing Oncohistone Mutants Using Yeast as a Model
 , ,
, , 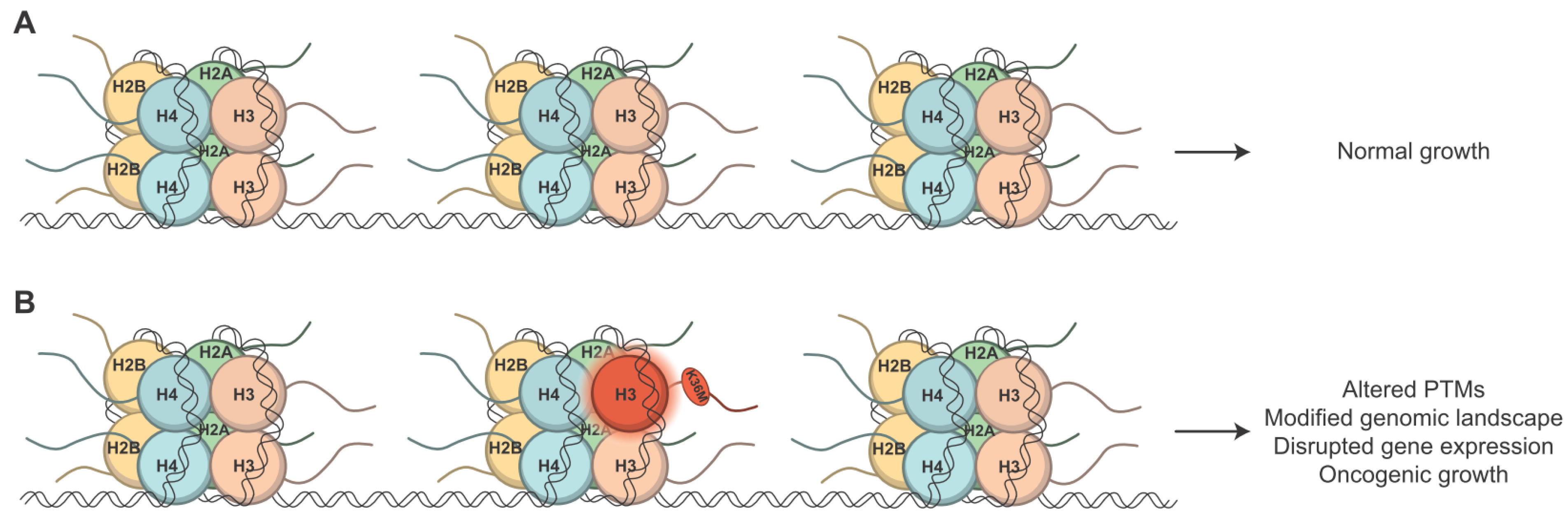{kind=link}
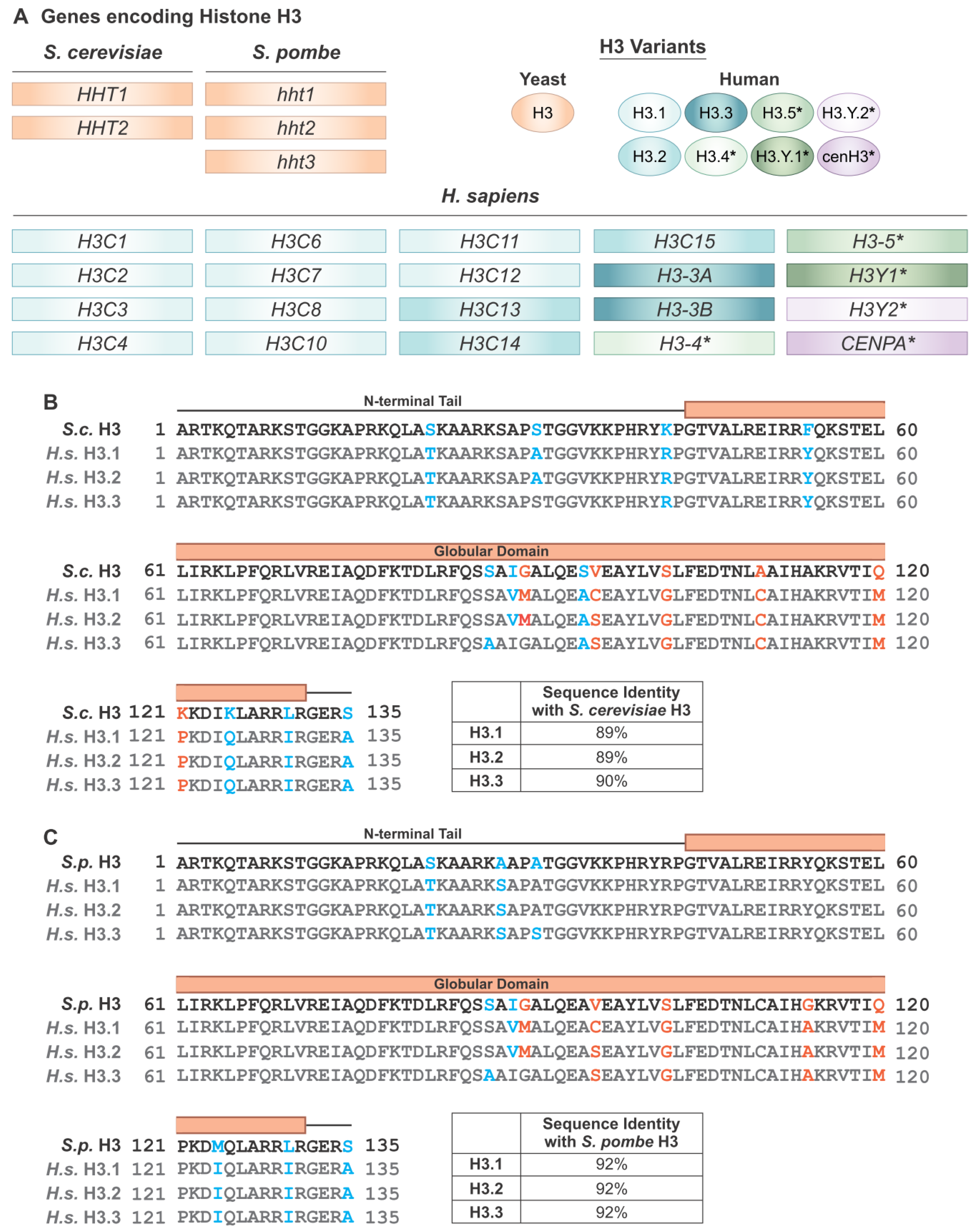{kind=link}
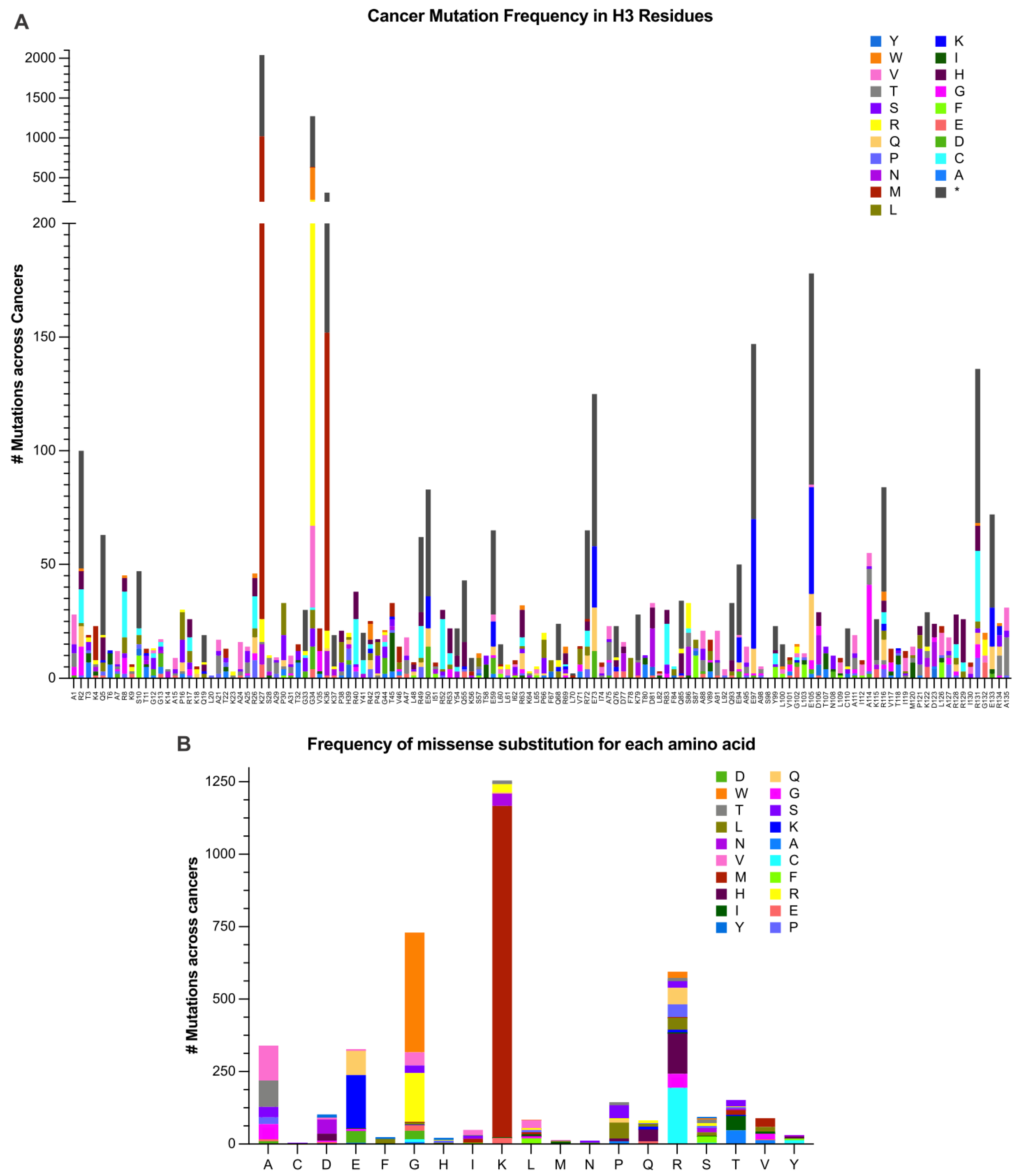{kind=link}
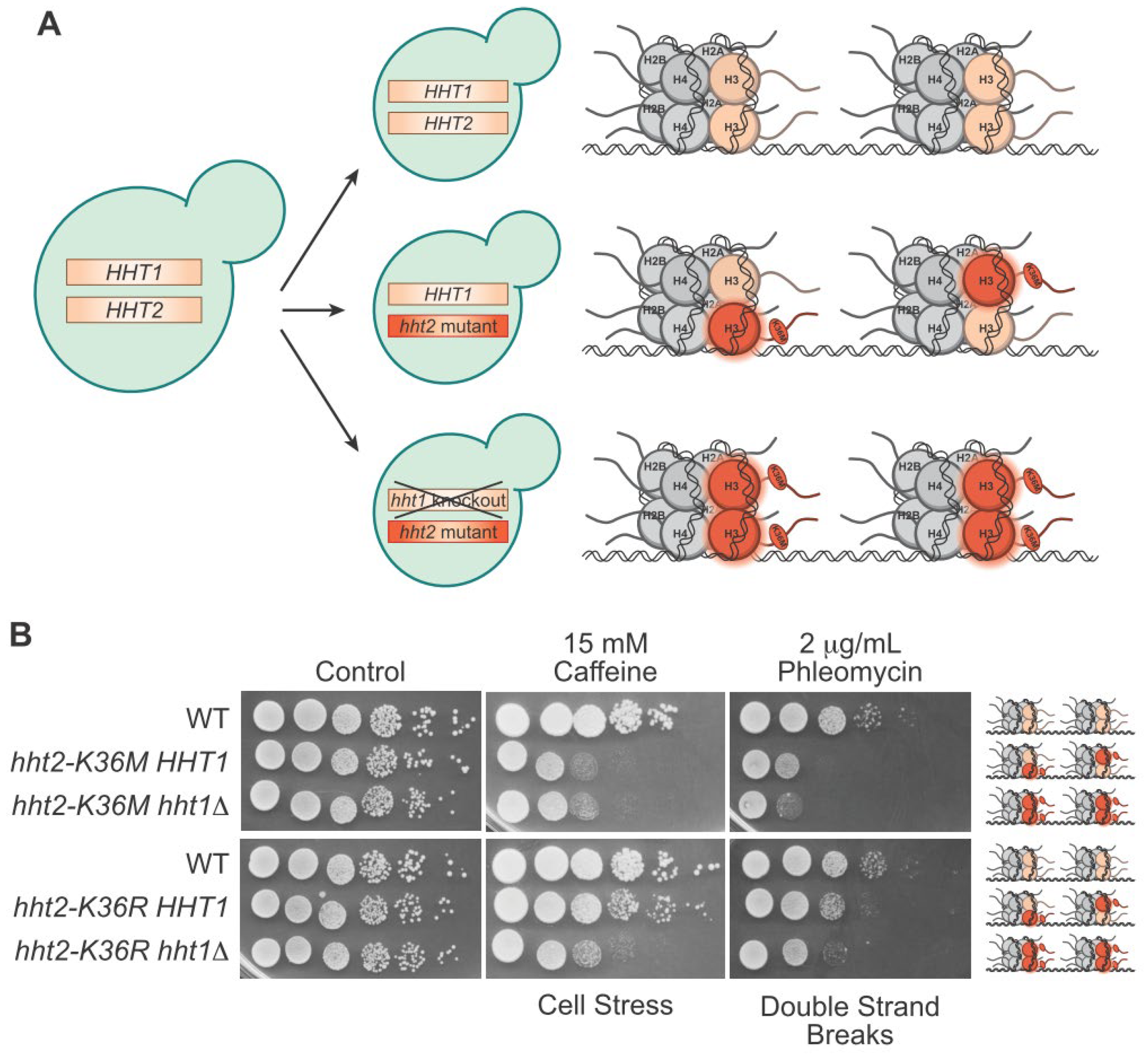{kind=link}
Abstract
:1. Introduction
2. Histones and Nucleosomes in Humans and Yeast
3. Oncohistones Are Novel, Cancer-Causing Histone Mutants
4. Advantages of Using Yeast Models to Investigate Oncohistones
5. Progress towards Histone and Oncohistone Characterization via Yeast Models
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, J.; Murphy, S.L.; Kochanek, K.D.; Arias, E. Mortality in the United States; 2021 NCHS Data Brief; National Center for Health Statistics: Hyattsville, MD, USA, 2022; pp. 1–8.
- Hartwell, L.H. Yeast and cancer. Biosci. Rep. 2004, 24, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M. Histone structure and function. Curr. Opin. Cell Biol. 1991, 3, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Millar, C.B.; Grunstein, M. Genome-wide patterns of histone modifications in yeast. Nat. Rev. Mol. Cell Biol. 2006, 7, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Dechassa, M.L.; Tremethick, D.J. New insights into nucleosome and chromatin structure: An ordered state or a disordered affair? Nat. Rev. Mol. Cell Biol. 2012, 13, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Stützer, A.; Liokatis, S.; Kiesel, A.; Schwarzer, D.; Sprangers, R.; Söding, J.; Selenko, P.; Fischle, W. Modulations of DNA Contacts by Linker Histones and Post-translational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol. Cell 2016, 61, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Albers, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P.; Wilson, J.; Hunt, T. Molecular Biology of the Cell, 5th ed.; Garland Science: New York, NY, USA, 2008. [Google Scholar]
- Flaus, A.; Downs, J.A.; Owen-Hughes, T. Histone isoforms and the oncohistone code. Curr. Opin. Genet. Dev. 2021, 67, 61–66. [Google Scholar] [CrossRef]
- Seal, R.L.; Denny, P.; Bruford, E.A.; Gribkova, A.K.; Landsman, D.; Marzluff, W.F.; McAndrews, M.; Panchenko, A.R.; Shaytan, A.K.; Talbert, P.B. A standardized nomenclature for mammalian histone genes. Epigenetics Chromatin 2022, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bassett, E.; Chakravarti, A.; Parthun, M.R. Replication-dependent histone isoforms: A new source of complexity in chromatin structure and function. Nucleic Acids Res. 2018, 46, 8665–8678. [Google Scholar] [CrossRef]
- McBurney, K.L.; Leung, A.; Choi, J.K.; Martin, B.J.; Irwin, N.A.; Bartke, T.; Nelson, C.J.; Howe, L.J. Divergent Residues Within Histone H3 Dictate a Unique Chromatin Structure in Saccharomyces cerevisiae. Genetics 2016, 202, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Amigo, R.; Farkas, C.; Gidi, C.; Hepp, M.I.; Cartes, N.; Tarifeño, E.E.; Workman, J.L.; Gutiérrez, J.L. The linker histone Hho1 modulates the activity of ATP-dependent chromatin remodeling complexes. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194781. [Google Scholar] [CrossRef] [PubMed]
- Libuda, D.E.; Winston, F. Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature 2006, 443, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.R.; Ganguli, D.; Nagarajavel, V.; Clark, D.J. Regulation of histone gene expression in budding yeast. Genetics 2012, 191, 7–20. [Google Scholar] [CrossRef]
- Smith, M.M.; Murray, K. Yeast H3 and H4 histone messenger RNAs are transcribed from two non-allelic gene sets. J. Mol. Biol. 1983, 169, 641–661. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Liu, Y.; Wang, A.; Qin, Y.; Luo, M.; Wu, Q.; Boeke, J.D.; Dai, J. Construction of Comprehensive Dosage-Matching Core Histone Mutant Libraries for Saccharomyces cerevisiae. Genetics 2017, 207, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.L.; Smith, M.M. Comparison of the structure and cell cycle expression of mRNAs encoded by two histone H3-H4 loci in Saccharomyces cerevisiae. Mol. Cell Biol. 1988, 8, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Burkhart, S.L.; Singh, R.K.; Kabbaj, M.H.; Gunjan, A. Histone dosage regulates DNA damage sensitivity in a checkpoint-independent manner by the homologous recombination pathway. Nucleic Acids Res. 2012, 40, 9604–9620. [Google Scholar] [CrossRef]
- Hereford, L.; Fahrner, K.; Woolford, J., Jr.; Rosbash, M.; Kaback, D.B. Isolation of yeast histone genes H2A and H2B. Cell 1979, 18, 1261–1271. [Google Scholar] [CrossRef]
- Norris, D.; Osley, M.A. The two gene pairs encoding H2A and H2B play different roles in the Saccharomyces cerevisiae life cycle. Mol. Cell Biol. 1987, 7, 3473–3481. [Google Scholar] [CrossRef]
- Hörz, W.; Zachau, H.G. Deoxyribonuclease II as a probe for chromatin structure. I. Location of cleavage sites. J. Mol. Biol. 1980, 144, 305–327. [Google Scholar] [CrossRef] [PubMed]
- Morse, R.H.; Pederson, D.S.; Dean, A.; Simpson, R.T. Yeast nucleosomes allow thermal untwisting of DNA. Nucleic Acids Res. 1987, 15, 10311–10330. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Suto, R.K.; Luger, K. Structure of the yeast nucleosome core particle reveals fundamental changes in internucleosome interactions. EMBO J. 2001, 20, 5207–5218. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta 2009, 1789, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Thorne, A.W.; Kmiciek, D.; Mitchelson, K.; Sautiere, P.; Crane-Robinson, C. Patterns of histone acetylation. Eur. J. Biochem. 1990, 193, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J. 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Schneider, J.; Bajwa, P.; Johnson, F.C.; Bhaumik, S.R.; Shilatifard, A. Rtt109 is required for proper H3K56 acetylation: A chromatin mark associated with the elongating RNA polymerase II. J. Biol. Chem. 2006, 281, 37270–37274. [Google Scholar] [CrossRef]
- Bernier, M.; Luo, Y.; Nwokelo, K.C.; Goodwin, M.; Dreher, S.J.; Zhang, P.; Parthun, M.R.; Fondufe-Mittendorf, Y.; Ottesen, J.J.; Poirier, M.G. Linker histone H1 and H3K56 acetylation are antagonistic regulators of nucleosome dynamics. Nat. Commun. 2015, 6, 10152. [Google Scholar] [CrossRef]
- Hong, L.; Schroth, G.P.; Matthews, H.R.; Yau, P.; Bradbury, E.M. Studies of the DNA binding properties of histone H4 amino terminus. Thermal denaturation studies reveal that acetylation markedly reduces the binding constant of the H4 “tail” to DNA. J. Biol. Chem. 1993, 268, 305–314. [Google Scholar] [CrossRef]
- Clayton, A.L.; Hazzalin, C.A.; Mahadevan, L.C. Enhanced histone acetylation and transcription: A dynamic perspective. Mol. Cell 2006, 23, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M.; O’Neill, L.P. Histone acetylation in chromatin and chromosomes. Semin. Cell Biol. 1995, 6, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.; Yue, W.W.; Oppermann, U.; Klose, R.J. Dynamic protein methylation in chromatin biology. Cell Mol. Life Sci. 2009, 66, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Miller, K.M. Histone methylation and the DNA damage response. Mutat. Res. Rev. Mutat. Res. 2019, 780, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gogol, M.; Carey, M.; Pattenden, S.G.; Seidel, C.; Workman, J.L. Infrequently transcribed long genes depend on the Set2/Rpd3S pathway for accurate transcription. Genes Dev. 2007, 21, 1422–1430. [Google Scholar] [CrossRef]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Humphrey, E.L.; Erlich, R.L.; Schneider, R.; Bouman, P.; Liu, J.S.; Kouzarides, T.; Schreiber, S.L. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. USA 2002, 99, 8695–8700. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Hong, T.; Walter, K.L.; Ewalt, M.; Michishita, E.; Hung, T.; Carney, D.; Peña, P.; Lan, F.; Kaadige, M.R.; et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 2006, 442, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Lam, U.T.F.; Tan, B.K.Y.; Poh, J.J.X.; Chen, E.S. Structural and functional specificity of H3K36 methylation. Epigenetics Chromatin 2022, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.K.; Nguyen, T.T.T.; Li, A.Y.; Yeo, Y.P.; Chen, E.S. Histone H3 lysine 36 methyltransferase mobilizes NER factors to regulate tolerance against alkylation damage in fission yeast. Nucleic Acids Res. 2018, 46, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.C.; Deegan, R.S.; Subramanian, L.; Gal, C.; Sarkar, S.; Blaikley, E.J.; Walker, C.; Hulme, L.; Bernhard, E.; Codlin, S.; et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun. 2014, 5, 4091. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, H.; Fong, N.; Erickson, B.; Bentley, D.L. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc. Natl. Acad. Sci. USA 2011, 108, 13564–13569. [Google Scholar] [CrossRef]
- Sorenson, M.R.; Jha, D.K.; Ucles, S.A.; Flood, D.M.; Strahl, B.D.; Stevens, S.W.; Kress, T.L. Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae. RNA Biol. 2016, 13, 412–426. [Google Scholar] [CrossRef]
- Yuan, H.; Li, N.; Fu, D.; Ren, J.; Hui, J.; Peng, J.; Liu, Y.; Qiu, T.; Jiang, M.; Pan, Q.; et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Investig. 2017, 127, 3375–3391. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef]
- Wiles, E.T.; Selker, E.U. H3K27 methylation: A promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017, 43, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Cheung, W.L.; Hsu, J.Y.; Diaz, R.L.; Smith, M.M.; Allis, C.D. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 2005, 120, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Sierra, F.; Lichtler, A.; Marashi, F.; Rickles, R.; Van Dyke, T.; Clark, S.; Wells, J.; Stein, G.; Stein, J. Organization of human histone genes. Proc. Natl. Acad. Sci. USA 1982, 79, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R. Sophisticated Conversations between Chromatin and Chromatin Remodelers, and Dissonances in Cancer. Int. J. Mol. Sci. 2021, 22, 5578. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSα interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef]
- Zhang, Y.; Shan, C.M.; Wang, J.; Bao, K.; Tong, L.; Jia, S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci. Rep. 2017, 7, 43906. [Google Scholar] [CrossRef]
- Shi, L.; Shi, J.; Shi, X.; Li, W.; Wen, H. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J. Mol. Biol. 2018, 430, 1562–1565. [Google Scholar] [CrossRef]
- Jiao, L.; Liu, X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015, 350, aac4383. [Google Scholar] [CrossRef]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.U.; Khazaei, S.; Marchione, D.M.; Lundgren, S.M.; Wang, X.; Weinberg, D.N.; Deshmukh, S.; Juretic, N.; Lu, C.; Allis, C.D.; et al. Histone H3.3 G34 mutations promote aberrant PRC2 activity and drive tumor progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27354–27364. [Google Scholar] [CrossRef] [PubMed]
- Mitchener, M.M.; Muir, T.W. Oncohistones: Exposing the nuances and vulnerabilities of epigenetic regulation. Mol. Cell 2022, 82, 2925–2938. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.L.; Bele, A.; Small, E.C.; Will, C.M.; Nabet, B.; Oyer, J.A.; Huang, X.; Ghosh, R.P.; Grzybowski, A.T.; Yu, T.; et al. A Mutation in Histone H2B Represents a New Class of Oncogenic Driver. Cancer Discov. 2019, 9, 1438–1451. [Google Scholar] [CrossRef]
- Fang, D.; Gan, H.; Lee, J.H.; Han, J.; Wang, Z.; Riester, S.M.; Jin, L.; Chen, J.; Zhou, H.; Wang, J.; et al. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science 2016, 352, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.R.; Yadav, R.K.; Henry, R.A.; Schreiner, P.; Matsuda, A.; Fernandez, A.G.; Finkelstein, D.; Campbell, M.; Kallappagoudar, S.; Jablonowski, C.M.; et al. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. Elife 2021, 10, e65369. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.B.; Kasper, L.H.; Fan, Y.; Jin, H.; Wu, G.; Shaw, T.I.; Zhu, X.; Larson, J.D.; Easton, J.; Shao, Y.; et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019, 137, 637–655. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef]
- Fnu, S.; Williamson, E.A.; De Haro, L.P.; Brenneman, M.; Wray, J.; Shaheen, M.; Radhakrishnan, K.; Lee, S.H.; Nickoloff, J.A.; Hromas, R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc. Natl. Acad. Sci. USA 2011, 108, 540–545. [Google Scholar] [CrossRef]
- Qiu, L.; Hu, X.; Jing, Q.; Zeng, X.; Chan, K.M.; Han, J. Mechanism of cancer: Oncohistones in action. J. Genet. Genom. 2018, 45, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Bonner, E.R.; Dawood, A.; Gordish-Dressman, H.; Eze, A.; Bhattacharya, S.; Yadavilli, S.; Mueller, S.; Waszak, S.M.; Nazarian, J. Pan-cancer atlas of somatic core and linker histone mutations. NPJ Genom. Med. 2023, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.Z.E.; Zhu, L.; Yang, D.; Ding, D.; Zhu, X.; Wan, Y.C.E.; Liu, J.; Ramakrishnan, S.; Chan, L.L.; Chan, S.Y.; et al. The elevated transcription of ADAM19 by the oncohistone H2BE76K contributes to oncogenic properties in breast cancer. J. Biol. Chem. 2021, 296, 100374. [Google Scholar] [CrossRef] [PubMed]
- Sankar, A.; Mohammad, F.; Sundaramurthy, A.K.; Wang, H.; Lerdrup, M.; Tatar, T.; Helin, K. Histone editing elucidates the functional roles of H3K27 methylation and acetylation in mammals. Nat. Genet. 2022, 54, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Côté, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Spedale, G.; Timmers, H.T.; Pijnappel, W.W. ATAC-king the complexity of SAGA during evolution. Genes Dev. 2012, 26, 527–541. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Xue, H.; Cao, M.; Bai, G.; Mu, Z.; Yao, Y.; Sun, S.; Fang, D.; Huang, J. Cryo-EM structure of SETD2/Set2 methyltransferase bound to a nucleosome containing oncohistone mutations. Cell Discov. 2021, 7, 32. [Google Scholar] [CrossRef]
- Lachner, M.; Sengupta, R.; Schotta, G.; Jenuwein, T. Trilogies of histone lysine methylation as epigenetic landmarks of the eukaryotic genome. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 209–218. [Google Scholar] [CrossRef]
- Su, W.P.; Hsu, S.H.; Chia, L.C.; Lin, J.Y.; Chang, S.B.; Jiang, Z.D.; Lin, Y.J.; Shih, M.Y.; Chen, Y.C.; Chang, M.S.; et al. Combined Interactions of Plant Homeodomain and Chromodomain Regulate NuA4 Activity at DNA Double-Strand Breaks. Genetics 2016, 202, 77–92. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Freitag, M. Histone Methylation by SET Domain Proteins in Fungi. Annu. Rev. Microbiol. 2017, 71, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Duina, A.A.; Miller, M.E.; Keeney, J.B. Budding yeast for budding geneticists: A primer on the Saccharomyces cerevisiae model system. Genetics 2014, 197, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Hyland, E.M.; Yuan, D.S.; Huang, H.; Bader, J.S.; Boeke, J.D. Probing nucleosome function: A highly versatile library of synthetic histone H3 and H4 mutants. Cell 2008, 134, 1066–1078. [Google Scholar] [CrossRef]
- Huang, H.; Maertens, A.M.; Hyland, E.M.; Dai, J.; Norris, A.; Boeke, J.D.; Bader, J.S. HistoneHits: A database for histone mutations and their phenotypes. Genome Res. 2009, 19, 674–681. [Google Scholar] [CrossRef]
- Nakanishi, S.; Sanderson, B.W.; Delventhal, K.M.; Bradford, W.D.; Staehling-Hampton, K.; Shilatifard, A.A. comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat. Struct. Mol. Biol. 2008, 15, 881–888. [Google Scholar] [CrossRef]
- Matsubara, K.; Sano, N.; Umehara, T.; Horikoshi, M. Global analysis of functional surfaces of core histones with comprehensive point mutants. Genes Cells 2007, 12, 13–33. [Google Scholar] [CrossRef]
- Collord, G.; Martincorena, I.; Young, M.D.; Foroni, L.; Bolli, N.; Stratton, M.R.; Vassiliou, G.S.; Campbell, P.J.; Behjati, S. Recurrent histone mutations in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2019, 184, 676–679. [Google Scholar] [CrossRef]
- Kuranda, K.; Leberre, V.; Sokol, S.; Palamarczyk, G.; François, J. Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol. Microbiol. 2006, 61, 1147–1166. [Google Scholar] [CrossRef]
- Chen, J.; Stubbe, J. Bleomycins: New methods will allow reinvestigation of old issues. Curr. Opin. Chem. Biol. 2004, 8, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Bagert, J.D.; Mitchener, M.M.; Patriotis, A.L.; Dul, B.E.; Wojcik, F.; Nacev, B.A.; Feng, L.; Allis, C.D.; Muir, T.W. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol. 2021, 17, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.M.; Boeke, J.D. Resetting the Yeast Epigenome with Human Nucleosomes. Cell 2017, 171, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Gaber, R.F. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell Biol. 1991, 11, 6317–6327. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, T.; Thireos, G. Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription. EMBO J. 1992, 11, 4145–4152. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.H.; Brownell, J.E.; Sobel, R.E.; Ranalli, T.A.; Cook, R.G.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 1996, 383, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Kruger, W.; Peterson, C.L.; Sil, A.; Coburn, C.; Arents, G.; Moudrianakis, E.N.; Herskowitz, I. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995, 9, 2770–2779. [Google Scholar] [CrossRef]
- Seol, J.H.; Kim, H.J.; Yoo, J.K.; Park, H.J.; Cho, E.J. Analysis of Saccharomyces cerevisiae histone H3 mutants reveals the role of the alphaN helix in nucleosome function. Biochem. Biophys. Res. Commun. 2008, 374, 543–548. [Google Scholar] [CrossRef]
- Ngubo, M.; Reid, J.L.; Patterton, H.G. Distinct structural groups of histone H3 and H4 residues have divergent effects on chronological lifespan in Saccharomyces cerevisiae. PLoS ONE 2022, 17, e0268760. [Google Scholar] [CrossRef]
- Yu, Y.; Srinivasan, M.; Nakanishi, S.; Leatherwood, J.; Shilatifard, A.; Sternglanz, R.A. conserved patch near the C terminus of histone H4 is required for genome stability in budding yeast. Mol. Cell Biol. 2011, 31, 2311–2325. [Google Scholar] [CrossRef] [PubMed]
- Lemon, L.D.; Kannan, S.; Mo, K.W.; Adams, M.; Choi, H.G.; Gulka, A.O.D.; Withers, E.S.; Nurelegne, H.T.; Gomez, V.; Ambrocio, R.E.; et al. A Saccharomyces cerevisiae model and screen to define the functional consequences of oncogenic histone missense mutations. G3 2022, 12, jkac120. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Eisen, A.; Gu, W.; Sattah, M.; Pannuti, A.; Zhou, J.; Cook, R.G.; Lucchesi, J.C.; Allis, C.D. ESA1 is a histone acetyltransferase that is essential for growth in yeast. Proc. Natl. Acad. Sci. USA 1998, 95, 3561–3565. [Google Scholar] [CrossRef] [PubMed]
- Cooke, S.L.; Soares, B.L.; Müller, C.A.; Nieduszynski, C.A.; Bastos de Oliveira, F.M.; de Bruin, R.A.M. Tos4 mediates gene expression homeostasis through interaction with HDAC complexes independently of H3K56 acetylation. J. Biol. Chem. 2021, 296, 100533. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 2017, 6, e27406. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, S.; Luger, K. Understanding histone acetyltransferase Rtt109 structure and function: How many chaperones does it take? Curr. Opin. Struct. Biol. 2011, 21, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.M.; Wang, J.; Xu, K.; Chen, H.; Yue, J.X.; Andrews, S.; Moresco, J.J.; Yates, J.R.; Nagy, P.L.; Tong, L.; et al. A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. eLife 2016, 5, e17903. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Fawwal, D.V.; Spangle, J.M.; Corbett, A.H.; Jones, C.Y. Exploring the Molecular Underpinnings of Cancer-Causing Oncohistone Mutants Using Yeast as a Model. J. Fungi 2023, 9, 1187. https://doi.org/10.3390/jof9121187
Zhang X, Fawwal DV, Spangle JM, Corbett AH, Jones CY. Exploring the Molecular Underpinnings of Cancer-Causing Oncohistone Mutants Using Yeast as a Model. Journal of Fungi. 2023; 9(12):1187. https://doi.org/10.3390/jof9121187
Chicago/Turabian StyleZhang, Xinran, Dorelle V. Fawwal, Jennifer M. Spangle, Anita H. Corbett, and Celina Y. Jones. 2023. "Exploring the Molecular Underpinnings of Cancer-Causing Oncohistone Mutants Using Yeast as a Model" Journal of Fungi 9, no. 12: 1187. https://doi.org/10.3390/jof9121187