
Data-Independent Acquisition (DIA) Is Superior for High Precision Phospho-Peptide Quantification in Magnaporthe oryzae


{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.1.1. Cultivation of Magnaporthe Oryzae
2.1.2. Cell Lysis and Protein Digest
2.1.3. Phospho-Peptide Enrichment
2.2. Peptide Identification
2.2.1. LC-MS/MS of M. oryzae Samples for Resource
2.2.2. LC-MS/MS of M. oryzae DIA Samples for Comparison
2.2.3. LC-MS/MS of M. oryzae DDA Samples for Comparison
2.2.4. Data Processing Parameters DIA
2.2.5. Data Processing Parameters DDA
2.2.6. Availability of Raw Data
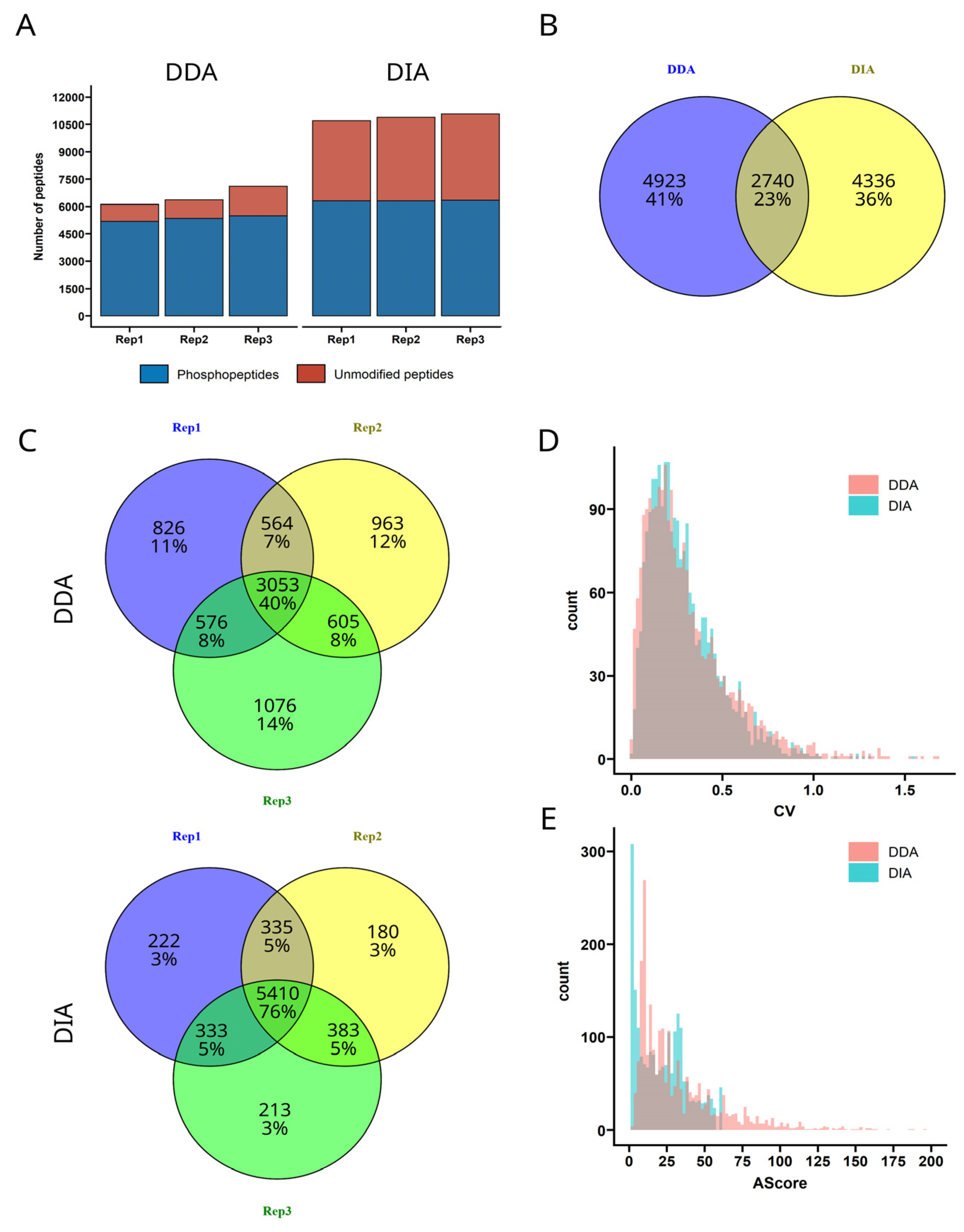
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Wilmanns, M.; Thornton, J.; Köhn, M. Elucidating human phosphatase-substrate networks. Sci. Signal. 2013, 6, rs10. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L. The crucial role of protein phosphorylation in cell signalingand its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Dengjel, J.; Akimov, V.; Olsen, J.V.; Bunkenborg, J.; Mann, M.; Blagoev, B.; Andersen, J.S. Quantitative proteomic assessment of very early cellular signaling events. Nat. Biotechnol. 2007, 25, 566–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigbolt, K.T.G.; Blagoev, B. Quantitative phosphoproteomics to characterize signaling networks. Semin. Cell Dev. Biol. 2012, 23, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kweon, H.K.; Andrews, P.C. Quantitative analysis of global phosphorylation changes with high-resolution tandem mass spectrometry and stable isotopic labeling. Methods 2013, 61, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Gao, C.; Kuster, B. On the potential of micro-flow LC-MS/MS in proteomics. Expert Rev. Proteom. 2022, 19, 153–164. [Google Scholar] [CrossRef]
- Wu, R.; Haas, W.; Dephoure, N.; Huttlin, E.L.; Zhai, B.; E Sowa, M.; Gygi, S.P. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat. Methods 2011, 8, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics revealswidespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Greening, D.W.; Kapp, E.A.; Simpson, R.J. The Peptidome Comes of Age: Mass Spectrometry-Based Characterization of the Circulating Cancer Peptidome. Enzymes 2017, 42, 27–64. [Google Scholar] [CrossRef]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef]
- Michna, T.; Tenzer, S. Quantitative Proteome and Phosphoproteome Profiling in Magnaporthe oryzae. Methods Mol. Biol. 2021, 2356, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Tariq, M.U.; Haseeb, M.; Aledhari, M.; Razzak, R.; Parizi, R.M.; Saeed, F. Methods for Proteogenomics Data Analysis, Challenges, and Scalability Bottlenecks: A Survey. IEEE Access 2021, 9, 5497–5516. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Hendrickson, R.C.; Pandey, A. Analysis of proteins and proteomes by mass spectrometry. Annu. Rev. Biochem. 2001, 70, 437–473. [Google Scholar] [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Bateman, N.W.; Goulding, S.P.; Shulman, N.J.; Gadok, A.K.; Szumlinski, K.K.; MacCoss, M.J.; Wu, C.C. Maximizing peptide identification events in proteomic workflows using data-dependent acquisition (DDA). Mol. Cell. Proteom. 2014, 13, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wen, Z.; Washburn, M.P.; Florens, L. Effect of dynamic exclusion duration on spectral count based quantitative proteomics. Anal. Chem. 2009, 81, 6317–6326. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Chen, X.; Zhong, L.; Yin, Y. Development of data-independent acquisition workflows for metabolomic analysis on a quadrupole-orbitrap platform. Talanta 2017, 164, 128–136. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, L.; Shan, B.; Chen, W.; Xie, M.; Yuen, D.; Zhang, W.; Zhang, Z.; Lajoie, G.A.; Ma, B. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol. Cell. Proteom. 2012, 11, M111.010587. [Google Scholar] [CrossRef] [Green Version]
- Nys, G.; Nix, C.; Cobraiville, G.; Servais, A.C.; Fillet, M. Enhancing protein discoverability by data independent acquisition assisted by ion mobility mass spectrometry. Talanta 2020, 213, 120812. [Google Scholar] [CrossRef]
- Li, K.W.; Gonzalez-Lozano, M.A.; Koopmans, F.; Smit, A.B. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Front. Mol. Neurosci. 2020, 13, 248. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Huang, P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics 2021, 17, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2019, 17, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Barkovits, K.; Pacharra, S.; Pfeiffer, K.; Steinbach, S.; Eisenacher, M.; Marcus, K.; Uszkoreit, J. Reproducibility, specificity and accuracy of relative quantification using spectral librarybased data-independent acquisition. Mol. Cell. Proteom. 2020, 19, 181–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnert, S.; Heck, L.; Gruber, C.; Neumann, H.; Distler, U.; Tenzer, S.; Yemelin, A.; Thines, E.; Jacob, S. Fungicide resistance toward fludioxonil conferred by overexpression of the phosphatase gene MoPTP2 in Magnaporthe oryzae. Mol. Microbiol. 2019, 111, 662–677. [Google Scholar] [CrossRef]
- Bohnert, S.; Antelo, L.; Grünewald, C.; Yemelin, A.; Andresen, K.; Jacob, S. Rapid adaptation of signaling networks in the fungal pathogen Magnaporthe oryzae. BMC Genom. 2019, 20, 763. [Google Scholar] [CrossRef] [Green Version]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [Green Version]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Bernhardt, O.M.; Hogrebe, A.; Martinez-Val, A.; Verbeke, L.; Gandhi, T.; Kelstrup, C.D.; Reiter, L.; Olsen, J.V. Rapid and site-specific deep phosphoproteome profiling by data-independent acquisition without the need for spectral libraries. Nat. Commun. 2020, 11, 787. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Val, A.; Bekker-Jensen, D.B.; Hogrebe, A.; Olsen, J.V. Data Processing and Analysis for DIA-Based Phosphoproteomics Using Spectronaut. Methods Mol. Biol. 2021, 2361, 95–107. [Google Scholar] [CrossRef]
- Kitata, R.B.; Choong, W.-K.; Tsai, C.-F.; Lin, P.-Y.; Chen, B.-S.; Chang, Y.-C.; Nesvizhskii, A.I.; Sung, T.-Y.; Chen, Y.-J. A data-independent acquisition-based global phosphoproteomics system enables deep profiling. Nat. Commun. 2021, 12, 2539. [Google Scholar] [CrossRef] [PubMed]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Xuan, Y.; Sondermann, J.; Schmidt, M.; Gomez-Varela, D.; Reiter, L. Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol. Cell. Proteom. 2017, 16, 2296–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, W.L.; Gokce, E.; Randall, S.M.; Oh, Y.; Eyre, A.; Muddiman, D.C.; Dean, R.A. Phosphoproteome Analysis Links Protein Phosphorylation to Cellular Remodeling and Metabolic Adaptation during Magnaporthe oryzae Appressorium Development. J. Proteome Res. 2015, 14, 2408–2424. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bersching, K.; Michna, T.; Tenzer, S.; Jacob, S. Data-Independent Acquisition (DIA) Is Superior for High Precision Phospho-Peptide Quantification in Magnaporthe oryzae. J. Fungi 2023, 9, 63. https://doi.org/10.3390/jof9010063
Bersching K, Michna T, Tenzer S, Jacob S. Data-Independent Acquisition (DIA) Is Superior for High Precision Phospho-Peptide Quantification in Magnaporthe oryzae. Journal of Fungi. 2023; 9(1):63. https://doi.org/10.3390/jof9010063
Chicago/Turabian StyleBersching, Katharina, Thomas Michna, Stefan Tenzer, and Stefan Jacob. 2023. "Data-Independent Acquisition (DIA) Is Superior for High Precision Phospho-Peptide Quantification in Magnaporthe oryzae" Journal of Fungi 9, no. 1: 63. https://doi.org/10.3390/jof9010063