
Recent Advances in Directed Yeast Genome Evolution

Abstract
:1. Introduction
2. DNA-Replication Protein-Related Methods
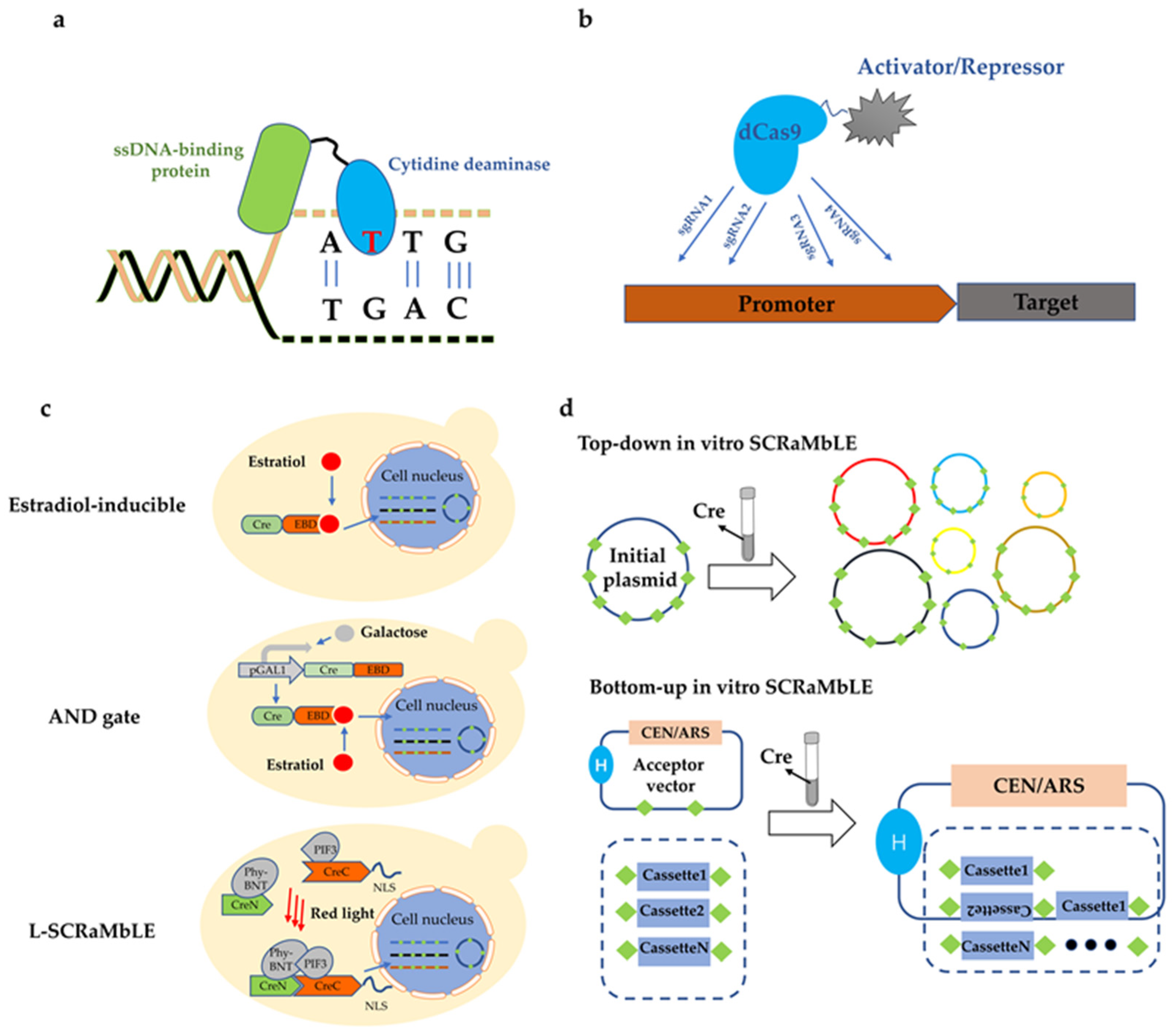
3. CRISPR/Cas9 Driving Genome Evolutionary Engineering
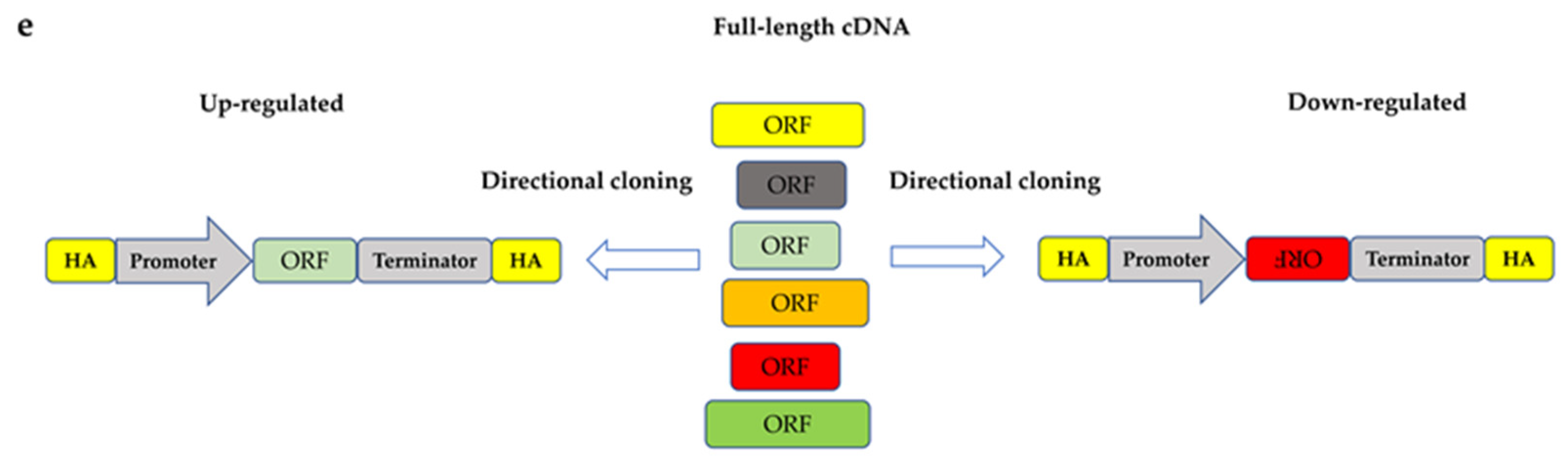
3.1. Combiantions of Up- and Downregulations
3.2. SgRNA Design

{kind=link}
{kind=link}
| Name | Description | gRNA | Applications | Reference |
|---|---|---|---|---|
| CRISPRi | dCas12a-Mxi1 | Targeting heterologous β-carotene biosynthesis pathway genes crtE, crtYB, crtI | β-carotene production | [43] |
| dCas9 | Targeting PFK1 and PYK1 | N-acetylglucosamine production | [44] | |
| dCas9 | Targeting seven genes in branch pathways of β-amyrin production | β-amyrin production | [45] | |
| dCas9-Mxi1 | Targeting over 98% of essential and respiratory growth-essential genes | Acetic-acid Tolerance | [46] | |
| dCas9-Mxi1 | Targeting transcription start site (TSS) in genome scale | Mining of haploinsufficient genes and identification of adenine and arginine biosynthesis genes | [47] | |
| dCas9-Mxi1 | Targeting 161 transcriptional factors and 129 protein kinase | Growth in lignocellulose hydrolysate | [40] | |
| CRISPRa | dCas9-VP64 | Targeting 52 genes | Thermotolerance | [48] |
| CHAnGE | Global deletion of genes from genome by homologous recombination via gRNA and donor synthesized on chip | Synthesized on chip in ~100% gene coverage | Acetic acid and furfural tolerance | [37] |
| STEPS | Combination of dCas9-Mix1 and dCas9-VPR to simultaneously up- and downregulate transcriptional levels, respectively | Graded targeting of genes involved in glycerol production, PPP genes for 3-DHS production and xylose catabolism | Glycerol and 3-DHS production, xylose catabolism | [38] |
| CRSPRi/a | Cas9-VPR | Four genes HMG1, ERG9, DPP1, and UPC2 | α-santalene production | [42] |
| CRSPRi/a | dCas9 or dCas9-Mxi1dCas9-VP64 or dCas9-VPR | Targeting four genes HRK1, SSK2, ISC1 and BDH2 | Tolerance towards lignocellulosic hydrolysate. | [49] |
| MAGIC | Combination of dLbCas12a-VP, dSpCas9-RD1152 and SaCas9 to simultaneously upregulate, downregulate and delete genes, respectively | Synthesized on chip in ~100% gene coverage | GAL7 and HED1 expression levels | [39] |
4. SCRaMbLE Driving Yeast Chromosomal Rearrangement
4.1. Diversity
4.2. Controlling the Expression of Cre
4.3. In Vitro SCRaMbLE
5. Evolution in Transcriptional Levels
6. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Avalos, J.L.; Fink, G.R.; Stephanopoulos, G. Compartmentalization of metabolic pathways in yeast mitochondria improves the production of branched-chain alcohols. Nat. Biotechnol. 2013, 31, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, B.; Zhou, P.; Jiao, X.; Yao, Z.; Ye, L.; Yu, H. Fermentative production of Vitamin E tocotrienols in Saccharomyces cerevisiae under cold-shock-triggered temperature control. Nat. Commun. 2020, 11, 5155. [Google Scholar] [CrossRef] [PubMed]
- Meadows, A.L.; Hawkins, K.M.; Tsegaye, Y.; Antipov, E.; Kim, Y.; Raetz, L.; Dahl, R.H.; Tai, A.; Mahatdejkul-Meadows, T.; Xu, L.; et al. Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature 2016, 537, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Lv, X.; Ye, L.; Zhou, P.; Yu, H. Construction of lycopene-overproducing Saccharomyces cerevisiae by combining directed evolution and metabolic engineering. Metab. Eng. 2015, 30, 69–78. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, C.; Sun, W.; Zhou, A.; Wang, Y.; Zhang, G.; Zhou, X.; Huo, Y.; Li, C. Boosting 11-oxo-β-amyrin and glycyrrhetinic acid synthesis in Saccharomyces cerevisiae via pairing novel oxidation and reduction system from legume plants. Metab. Eng. 2018, 45, 43–50. [Google Scholar] [CrossRef]
- Yu, T.; Zhou, Y.J.; Huang, M.; Liu, Q.; Pereira, R.; David, F.; Nielsen, J. Reprogramming Yeast Metabolism from Alcoholic Fermentation to Lipogenesis. Cell 2018, 174, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Huang, M.; Chen, Y.; Siewers, V.; Nielsen, J. Global rewiring of cellular metabolism renders Saccharomyces cerevisiae Crabtree negative. Nat. Commun. 2018, 9, 3059. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, T.E.; Salazar, M.J.; Weng, L.L.; Palsson, B.O.; Feist, A.M. The emergence of adaptive laboratory evolution as an efficient tool for biological discovery and industrial biotechnology. Metab. Eng. 2019, 56, 1–16. [Google Scholar] [CrossRef]
- Cao, M.; Tran, V.G.; Zhao, H. Unlocking nature’s biosynthetic potential by directed genome evolution. Curr. Opin. Biotechnol. 2020, 66, 95–104. [Google Scholar] [CrossRef]
- Simon, M.; Giot, L.; Faye, G. The 3′ to 5′ exonuclease activity located in the DNA polymerase delta subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J. 1991, 10, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Swan, M.K.; Johnson, R.E.; Prakash, L.; Prakash, S.; Aggarwal, A.K. Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat. Struct. Mol. Biol. 2009, 16, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kram Er, W.; Kram Er, B.; Williamson, M.S.; Fogel, S. Cloning and nucleotide sequence of DNA mismatch repair gene PMS1 from Saccharomyces cerevisiae: Homology of PMS1 to procaryotic MutL and HexB. J. Bacteriol. 1989, 171, 5339–5346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, A.J.; Kennedy, S.R.; Knowels, G.M.; Schultz, E.M.; Preston, B.D. DNA replication error-induced extinction of diploid yeast. Genetics 2014, 196, 677–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, A.; Johnson, A.L.; Johnston, L.H.; Sugino, A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993, 12, 1467–1473. [Google Scholar] [CrossRef]
- Fortune, J.M.; Pavlov, Y.I.; Welch, C.M.; Johansson, E.; Burgers, P.M.; Kunkel, T.A. Saccharomyces cerevisiae DNA polymerase delta: High fidelity for base substitutions but lower fidelity for single- and multi-base deletions. J. Biol. Chem. 2005, 280, 29980–29987. [Google Scholar] [CrossRef] [Green Version]
- Pursell, Z.F.; Isoz, I.; Lundstrom, E.B.; Johansson, E.; Kunkel, T.A. Yeast DNA Polymerase ε Participates in Leading-Strand DNA Replication. Science 2007, 317, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Flood, C.L.; Rodriguez, G.P.; Bao, G.; Shockley, A.H.; Kow, Y.W.; Crouse, G.F. Replicative DNA polymerase delta but not epsilon proofreads errors in Cis and in Trans. PLoS Genet. 2015, 11, e1005049. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, C.; Itadani, A.; Sugino, A.; Furusawa, M. Isolation of thermotolerant mutants by using proofreading-deficient DNA polymerase delta as an effective mutator in Saccharomyces cerevisiae. Genes. Genet. Syst. 2006, 81, 391–397. [Google Scholar] [CrossRef] [Green Version]
- Fukui, K. DNA mismatch repair in eukaryotes and bacteria. J. Nucleic. Acids. 2010, 2010, 260512. [Google Scholar] [CrossRef] [Green Version]
- Suter, C.M.; Martin, D.I.K.; Ward, R.L. Germline epimutation of MLH1 in individuals with multiple cancers. Nat. Genet. 2004, 36, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.S.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Leach, F.; Nicolai, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomaki, P.; Sistonen, P.; Aaltonen, L.; Nystromlahti, M. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Zhai, J.; Hingorani, M.M. Saccharomyces cerevisiae Msh2-Msh6 DNA binding kinetics reveal a mechanism of targeting sites for DNA mismatch repair. Proc. Natl. Acad. Sci. USA 2010, 107, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, B.; Kramer, W.; Williamson, M.S.; Fogel, S. Heteroduplex DNA correction in Saccharomyces cerevisiae is mismatch specific and requires functional PMS genes. Mol. Cell. Biol. 1989, 9, 4432–4440. [Google Scholar]
- White, J.H.; Lusnak, K.; Fogel, S. Mismatch-specific post-meiotic segregation frequency in yeast suggests a heteroduplex recombination intermediate. Nature 1985, 315, 350–352. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y.; et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Zhao, D.; Li, J.; Li, S.; Xin, X.; Hu, M.; Price, M.A.; Rosser, S.J.; Bi, C.; Zhang, X. Glycosylase base editors enable C-to-A and C-to-G base changes. Nat. Biotechnol. 2021, 39, 35–40. [Google Scholar] [CrossRef]
- Pan, Y.; Xia, S.; Dong, C.; Pan, H.; Cai, J.; Huang, L.; Xu, Z.; Lian, J. Random Base Editing for Genome Evolution in Saccharomyces cerevisiae. ACS Synth. Biol. 2021, 10, 2440–2446. [Google Scholar] [CrossRef]
- Hall, M.C.; Matson, S.W. Helicase motifs: The engine that powers DNA unwinding. Mol. Microbiol. 1999, 34, 867–877. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, D.; Li, J.; Hu, M.; Xin, X.; Price, M.A.; Li, Q.; Liu, L.; Li, S.; Rosser, S.J.; et al. Helicase-AID: A novel molecular device for base editing at random genomic loci. Metab. Eng. 2021, 67, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzadfard, F.; Perli, S.D.; Lu, T.K. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol. 2013, 2, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Z.; HamediRad, M.; Xue, P.; Xiao, H.; Tasan, I.; Chao, R.; Liang, J.; Zhao, H. Genome-scale engineering of Saccharomyces cerevisiae with single-nucleotide precision. Nat. Biotechnol. 2018, 36, 505–508. [Google Scholar] [CrossRef]
- Deaner, M.; Alper, H.S. Systematic testing of enzyme perturbation sensitivities via graded dCas9 modulation in Saccharomyces cerevisiae. Metab. Eng. 2017, 40, 14–22. [Google Scholar] [CrossRef]
- Lian, J.; Schultz, C.; Cao, M.; HamediRad, M.; Zhao, H. Multi-functional genome-wide CRISPR system for high throughput genotype-phenotype mapping. Nat. Commun. 2019, 10, 5794. [Google Scholar] [CrossRef]
- Gutmann, F.; Jann, C.; Pereira, F.; Johansson, A.; Steinmetz, L.M.; Patil, K.R. CRISPRi screens reveal genes modulating yeast growth in lignocellulose hydrolysate. Biotechnol. Biofuels 2021, 14, 41. [Google Scholar] [CrossRef]
- Roy, K.R.; Smith, J.D.; Vonesch, S.C.; Lin, G.; Tu, C.S.; Lederer, A.R.; Chu, A.; Suresh, S.; Nguyen, M.; Horecka, J.; et al. Multiplexed precision genome editing with trackable genomic barcodes in yeast. Nat. Biotechnol. 2018, 36, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Jiang, L.; Xu, S.; Huang, L.; Cai, J.; Lian, J.; Xu, Z. A Single Cas9-VPR Nuclease for Simultaneous Gene Activation, Repression, and Editing in Saccharomyces cerevisiae. ACS Synth. Biol. 2020, 9, 2252–2257. [Google Scholar] [CrossRef] [PubMed]
- Ciurkot, K.; Gorochowski, T.E.; Roubos, J.A.; Verwaal, R. Efficient multiplexed gene regulation in Saccharomyces cerevisiae using dCas12a. Nucleic Acids Res. 2021, 49, 7775–7790. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Lee, B.Y.; Oh, M.K. Combination of Three Methods to Reduce Glucose Metabolic Rate for Improving N-Acetylglucosamine Production in Saccharomyces cerevisiae. J. Agric. Food. Chem. 2018, 66, 13191–13198. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Zhang, G.; Qin, L.; Li, J.; Li, C. Simultaneously down-regulation of multiplex branch pathways using CRISPRi and fermentation optimization for enhancing beta-amyrin production in Saccharomyces cerevisiae. Synth. Syst. Biotechnol. 2019, 4, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, V.; Lind, U.; Onge, R.; Blomberg, A.; Nygrd, Y. A CRISPR Interference Screen of Essential Genes Reveals that Proteasome Regulation Dictates Acetic Acid Tolerance in Saccharomyces cerevisiae. mSystems 2021, 6, e00418-21. [Google Scholar] [CrossRef]
- Momen-Roknabadi, A.; Oikonomou, P.; Zegans, M.; Tavazoie, S. An inducible CRISPR interference library for genetic interrogation of Saccharomyces cerevisiae biology. Commun. Biol. 2020, 3, 723. [Google Scholar] [CrossRef]
- Li, P.; Fu, X.; Zhang, L.; Li, S. CRISPR/Cas-based screening of a gene activation library in Saccharomyces cerevisiae identifies a crucial role of OLE1 in thermotolerance. Microb. Biotechnol. 2019, 12, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Camara, E.; Lenitz, I.; Nygard, Y. A CRISPR activation and interference toolkit for industrial Saccharomyces cerevisiae strain KE6-12. Sci. Rep. 2020, 10, 14605. [Google Scholar] [CrossRef]
- Dymond, J.S.; Richardson, S.M.; Coombes, C.E.; Babatz, T.; Muller, H.; Annaluru, N.; Blake, W.J.; Schwerzmann, J.W.; Dai, J.; Lindstrom, D.L.; et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature 2011, 477, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Stracquadanio, G.; Wang, Y.; Yang, K.; Mitchell, L.A.; Xue, Y.; Cai, Y.; Chen, T.; Dymond, J.S.; Kang, K.; et al. SCRaMbLE generates designed combinatorial stochastic diversity in synthetic chromosomes. Genome Res. 2016, 26, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.J.; Wu, Y.; Yang, K.; Li, Y.; Xu, H.; Zhang, H.; Li, B.Z.; Li, X.; Xiao, W.H.; Zhou, X.; et al. Heterozygous diploid and interspecies SCRaMbLEing. Nat. Commun. 2018, 9, 1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blount, B.A.; Gowers, G.F.; Ho, J.C.H.; Ledesma-Amaro, R.; Jovicevic, D.; McKiernan, R.M.; Xie, Z.X.; Li, B.Z.; Yuan, Y.J.; Ellis, T. Rapid host strain improvement by in vivo rearrangement of a synthetic yeast chromosome. Nat. Commun. 2018, 9, 1932. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Wang, L.; Wang, Y.; Zhang, W.; Guo, Y.; Shen, Y.; Jiang, L.; Wu, Q.; Zhang, C.; Cai, Y.; et al. Identifying and characterizing SCRaMbLEd synthetic yeast using ReSCuES. Nat. Commun. 2018, 9, 1930. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wang, Y.; Chen, T.; Gao, F.; Gong, J.; Abramczyk, D.; Walker, R.; Zhao, H.; Chen, S.; Liu, W.; et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science 2017, 355, aaf4791. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Jin, J.; Han, M.-Z.; Li, B.-Z.; Yuan, Y.-J. Directed yeast genome evolution by controlled introduction of trans-chromosomic structural variations. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mercy, G.; Mozziconacci, J.; Scolari, V.F.; Yang, K.; Zhao, G.; Thierry, A.; Luo, Y.; Mitchell, L.A.; Shen, M.; Shen, Y.; et al. 3D organization of synthetic and scrambled chromosomes. Science 2017, 355, eaaf4597. [Google Scholar] [CrossRef] [Green Version]
- Richardson, S.M.; Mitchell, L.A.; Stracquadanio, G.; Yang, K.; Dymond, J.S.; Dicarlo, J.E.; Lee, D.; Huang, C.L.V.; Chandrasegaran, S.; Cai, Y.; et al. Design of a synthetic yeast genome. Science 2017, 355, 1040–1044. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Wu, Y.; Zhao, Y.; Zhang, Z.; Jiang, L.; Liu, L.; Zhang, Y.; Tang, J.; Yuan, Y.-J. Dynamics of synthetic yeast chromosome evolution shaped by hierarchical chromatin organization. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, J.; Xie, Z.X.; Ma, Y.; Chen, X.R.; Huang, Y.Q.; He, B.; Bin, J.; Li, B.Z.; Yuan, Y.J. Ring synthetic chromosome V SCRaMbLE. Nat. Commun. 2018, 9, 3783. [Google Scholar] [CrossRef]
- Jia, B.; Wu, Y.; Li, B.Z.; Mitchell, L.A.; Liu, H.; Pan, S.; Wang, J.; Zhang, H.R.; Jia, N.; Li, B.; et al. Precise control of SCRaMbLE in synthetic haploid and diploid yeast. Nat. Commun. 2018, 9, 1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochrein, L.; Mitchell, L.A.; Schulz, K.; Messerschmidt, K.; Mueller-Roeber, B. L-SCRaMbLE as a tool for light-controlled Cre-mediated recombination in yeast. Nat. Commun. 2018, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhu, R.Y.; Mitchell, L.A.; Ma, L.; Liu, R.; Zhao, M.; Jia, B.; Xu, H.; Li, Y.X.; Yang, Z.M.; et al. In vitro DNA SCRaMbLE. Nat. Commun. 2018, 9, 1935. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Luo, Z.; Wang, Y.; Pham, N.T.; Tuck, L.; Perez-Pi, I.; Liu, L.; Shen, Y.; French, C.; Auer, M.; et al. Rapid pathway prototyping and engineering using in vitro and in vivo synthetic genome SCRaMbLE-in methods. Nat. Commun. 2018, 9, 1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.S.; Lee, D.K.; Lee, H.; Lee, Y.; Jang, Y.S.; Kim, Y.H.; Yang, H.Y.; Lee, S.I.; Seol, W.; Kim, J.S. Phenotypic alteration of eukaryotic cells using randomized libraries of artificial transcription factors. Nat. Biotechnol. 2003, 21, 1208–1214. [Google Scholar] [CrossRef]
- Alper, H.; Moxley, J.; Nevoigt, E.; Fink, G.R.; Stephanopoulos, G. Engineering Yeast Transcription Machinery for Improved Ethanol Tolerance and Production. Science 2006, 314, 1565–1568. [Google Scholar] [CrossRef] [Green Version]
- Seong, Y.J.; Park, H.; Yang, J.; Kim, S.J.; Choi, W.; Kim, K.H.; Park, Y.C. Expression of a mutated SPT15 gene in Saccharomyces cerevisiae enhances both cell growth and ethanol production in microaerobic batch, fed-batch, and simultaneous saccharification and fermentations. Appl. Microbiol. Biotechnol. 2017, 101, 3567–3575. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, Y.; Guo, Y.; Wu, F.; Zhang, Y.; Qi, X.; Wang, Z.; Wang, Q. Stress tolerance enhancement via SPT15 base editing in Saccharomyces cerevisiae. Biotechnol. Biofuels 2021, 14, 155. [Google Scholar] [CrossRef]
- Si, T.; Chao, R.; Min, Y.; Wu, Y.; Ren, W.; Zhao, H. Automated multiplex genome-scale engineering in yeast. Nat. Commun. 2017, 8, 15187. [Google Scholar] [CrossRef]
- Li, P.; Fu, X.; Zhang, L.; Zhang, Z.; Li, J.; Li, S. The transcription factors Hsf1 and Msn2 of thermotolerant Kluyveromyces marxianus promote cell growth and ethanol fermentation of Saccharomyces cerevisiae at high temperatures. Biotechnol. Biofuels 2017, 10, 289. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.D.; Laloux, M.; Lehka, B.J.; Pedersen, L.E.; Jakociunas, T.; Jensen, M.K.; Keasling, J.D. A synthetic RNA-mediated evolution system in yeast. Nucleic Acids Res. 2021, 49, e88. [Google Scholar] [CrossRef] [PubMed]
- Crook, N.; Abatemarco, J.; Sun, J.; Wagner, J.M.; Schmitz, A.; Alper, H.S. In vivo continuous evolution of genes and pathways in yeast. Nat. Commun. 2016, 7, 13051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, E.M.; Muir, P.; Akhuetie-Oni, B.O.; Yellman, C.M.; Isaacs, F.J. Precise Editing at DNA Replication Forks Enables Multiplex Genome Engineering in Eukaryotes. Cell 2017, 171, 1453–1467. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Isaacs, F.J.; Carr, P.A.; Sun, Z.Z.; Xu, G.; Forest, C.R.; Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar] [CrossRef]
- Esvelt, K.M.; Carlson, J.C.; Liu, D.R. A system for the continuous directed evolution of biomolecules. Nature 2011, 472, 499–503. [Google Scholar] [CrossRef] [Green Version]
- DeBenedictis, E.A.; Chory, E.J.; Gretton, D.W.; Wang, B.; Golas, S.; Esvelt, K.M. Systematic molecular evolution enables robust biomolecule discovery. Nat. Methods 2022, 19, 55–64. [Google Scholar] [CrossRef]
- Zhong, Z.; Wong, B.G.; Ravikumar, A.; Arzumanyan, G.A.; Khalil, A.S.; Liu, C.C. Automated Continuous Evolution of Proteins in Vivo. ACS Synth. Biol. 2020, 9, 1270–1276. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Z.; Wang, Q.; Dai, Z. Recent Advances in Directed Yeast Genome Evolution. J. Fungi 2022, 8, 635. https://doi.org/10.3390/jof8060635
Yao Z, Wang Q, Dai Z. Recent Advances in Directed Yeast Genome Evolution. Journal of Fungi. 2022; 8(6):635. https://doi.org/10.3390/jof8060635
Chicago/Turabian StyleYao, Zhen, Qinhong Wang, and Zongjie Dai. 2022. "Recent Advances in Directed Yeast Genome Evolution" Journal of Fungi 8, no. 6: 635. https://doi.org/10.3390/jof8060635