
Morel Production Associated with Soil Nitrogen-Fixing and Nitrifying Microorganisms

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Statistical Analyses
3. Results
3.1. Sequencing Results
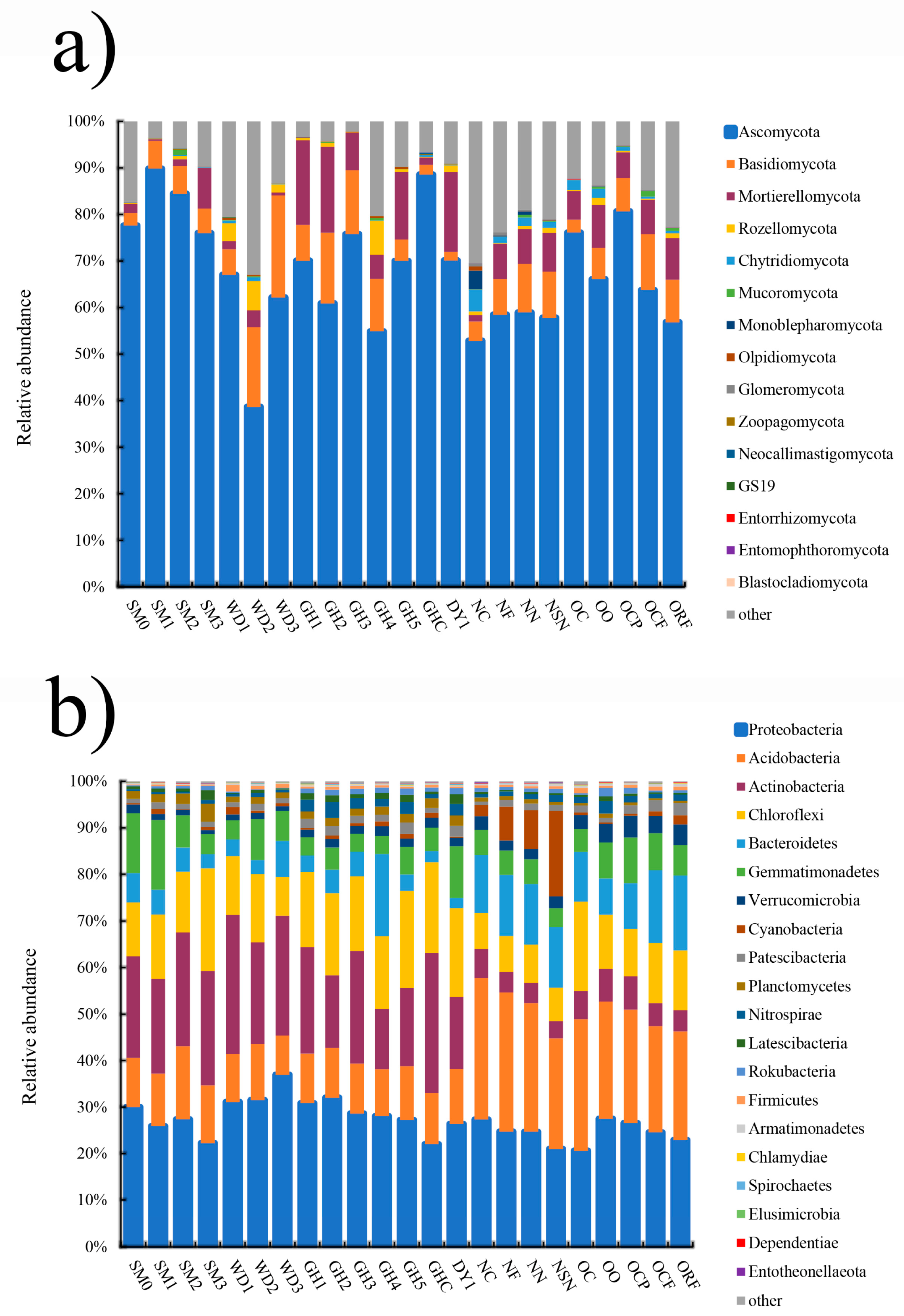
3.2. Microbial Community Composition and Structure Analysis in All Soil Samples
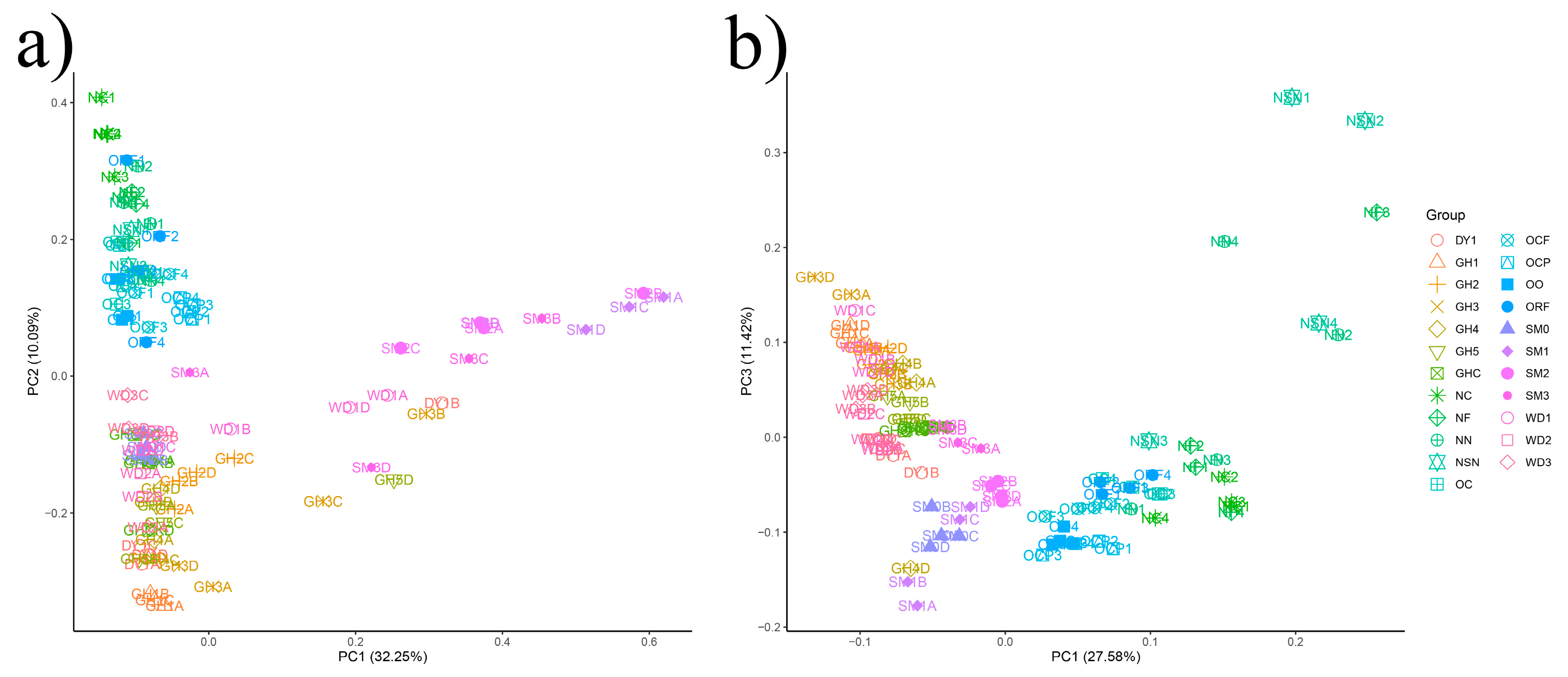
3.3. The Beta-Diversity of Soil Microbial Communities in All Samples
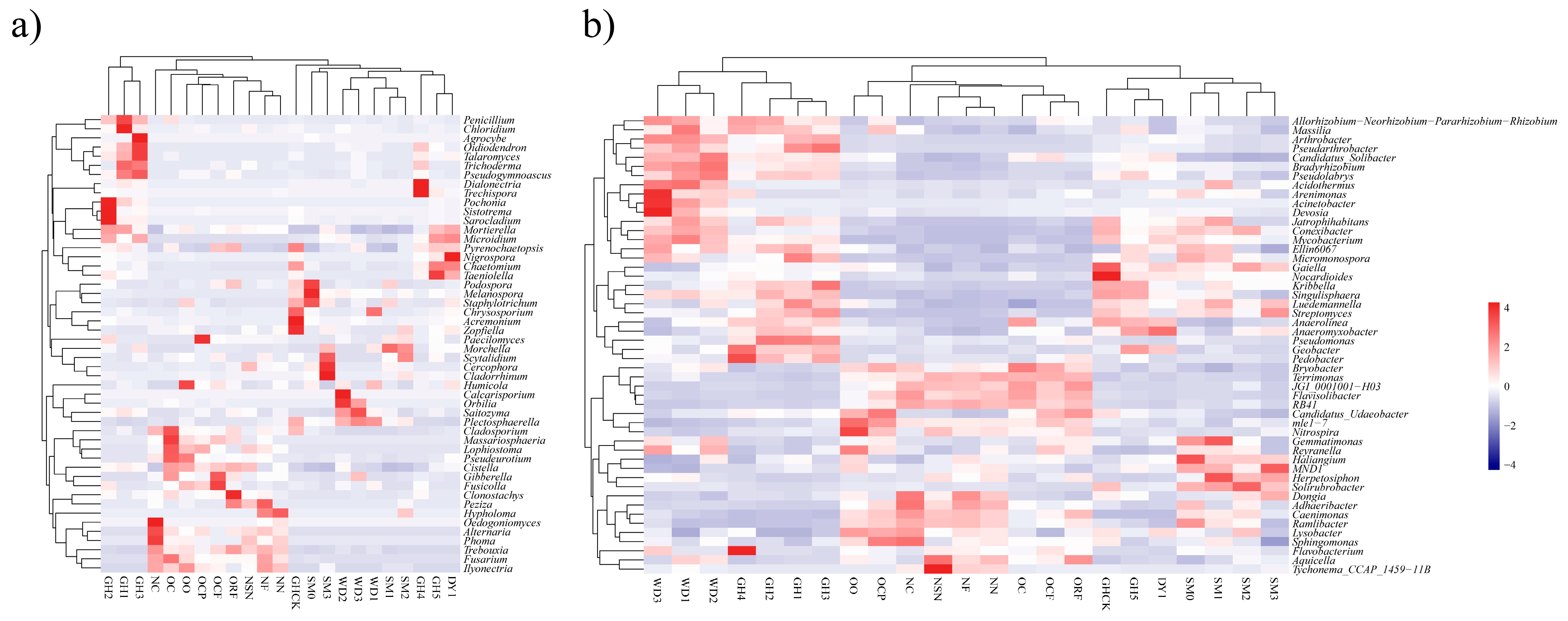
3.4. Heatmap Analyses of Soil Microbial Communities
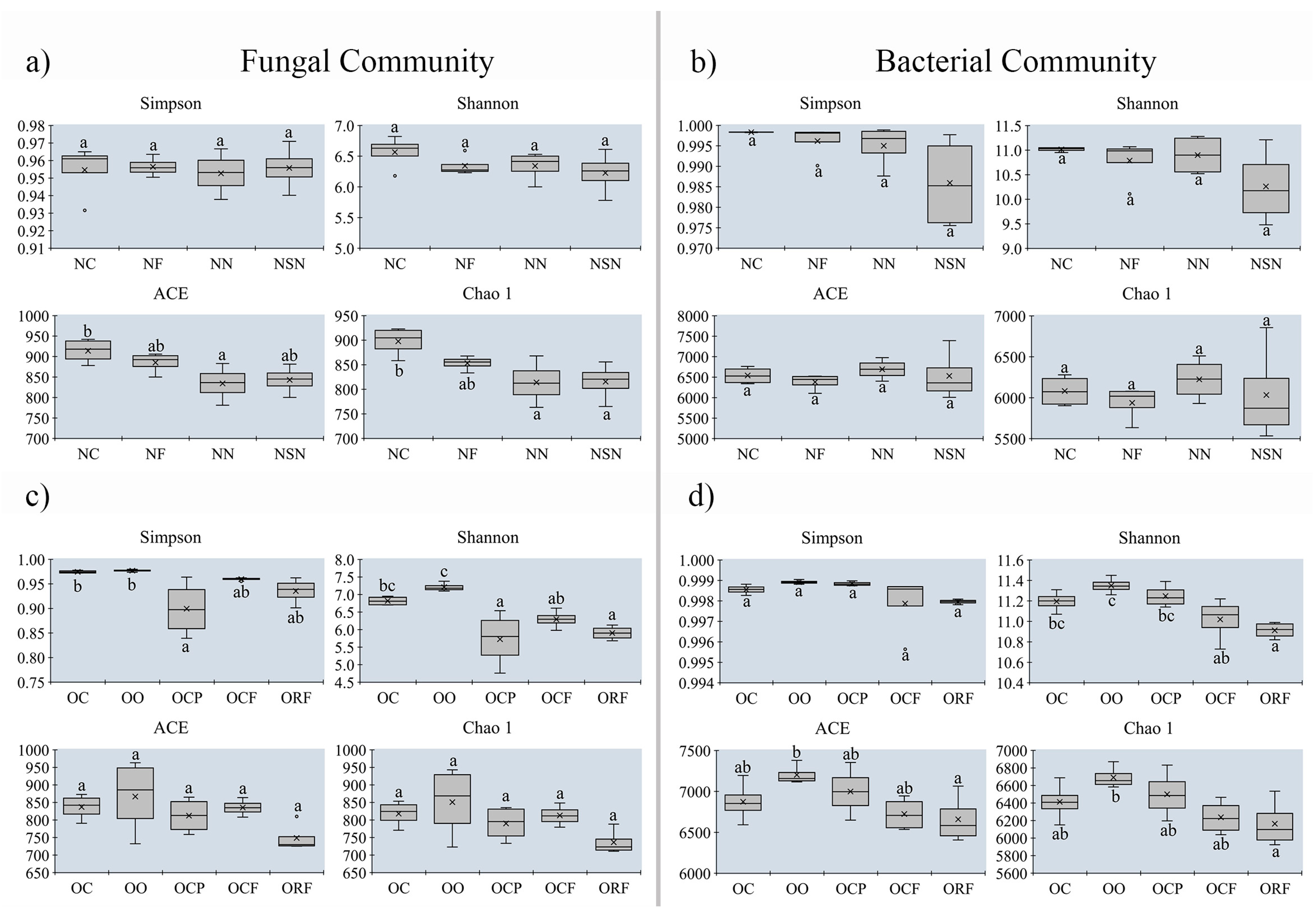
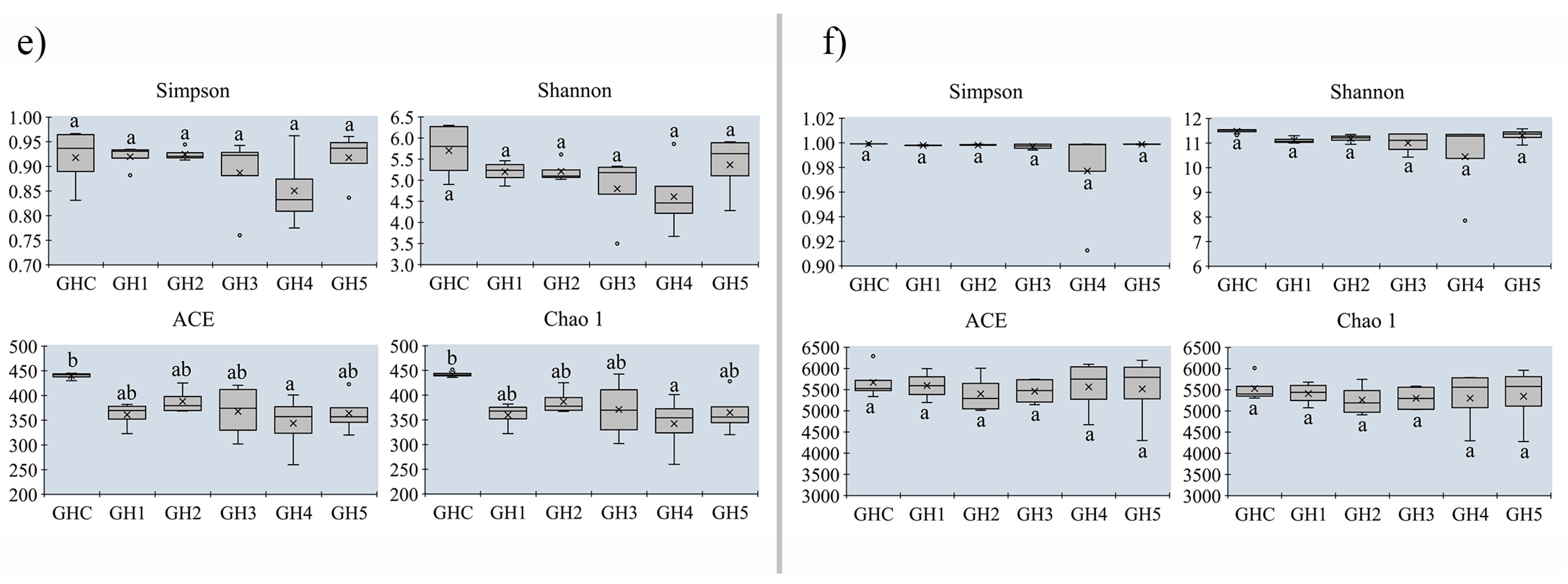
3.5. Changes in Alpha-Diversity on Soil Microbial Community between MUCS and MCS
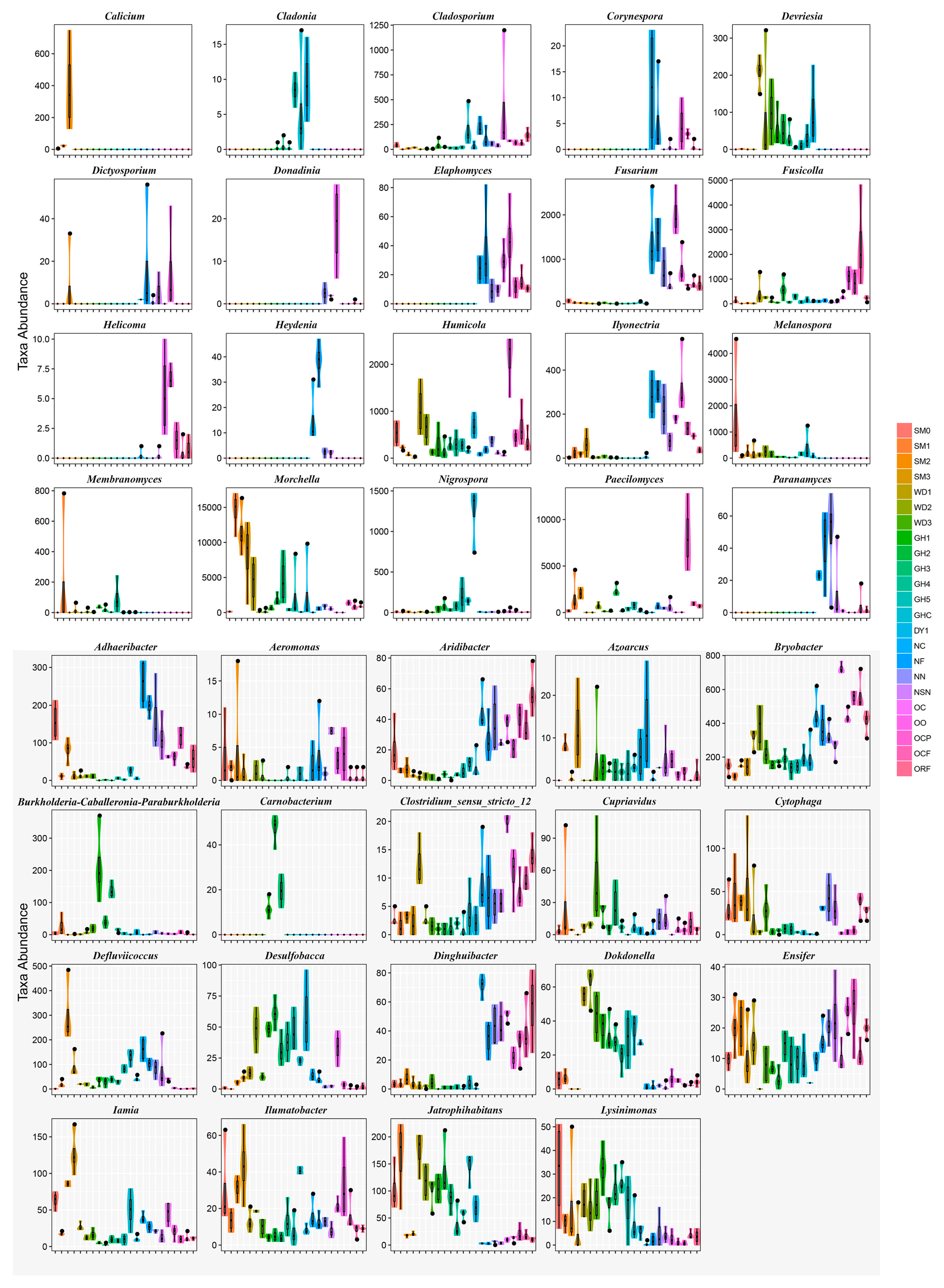
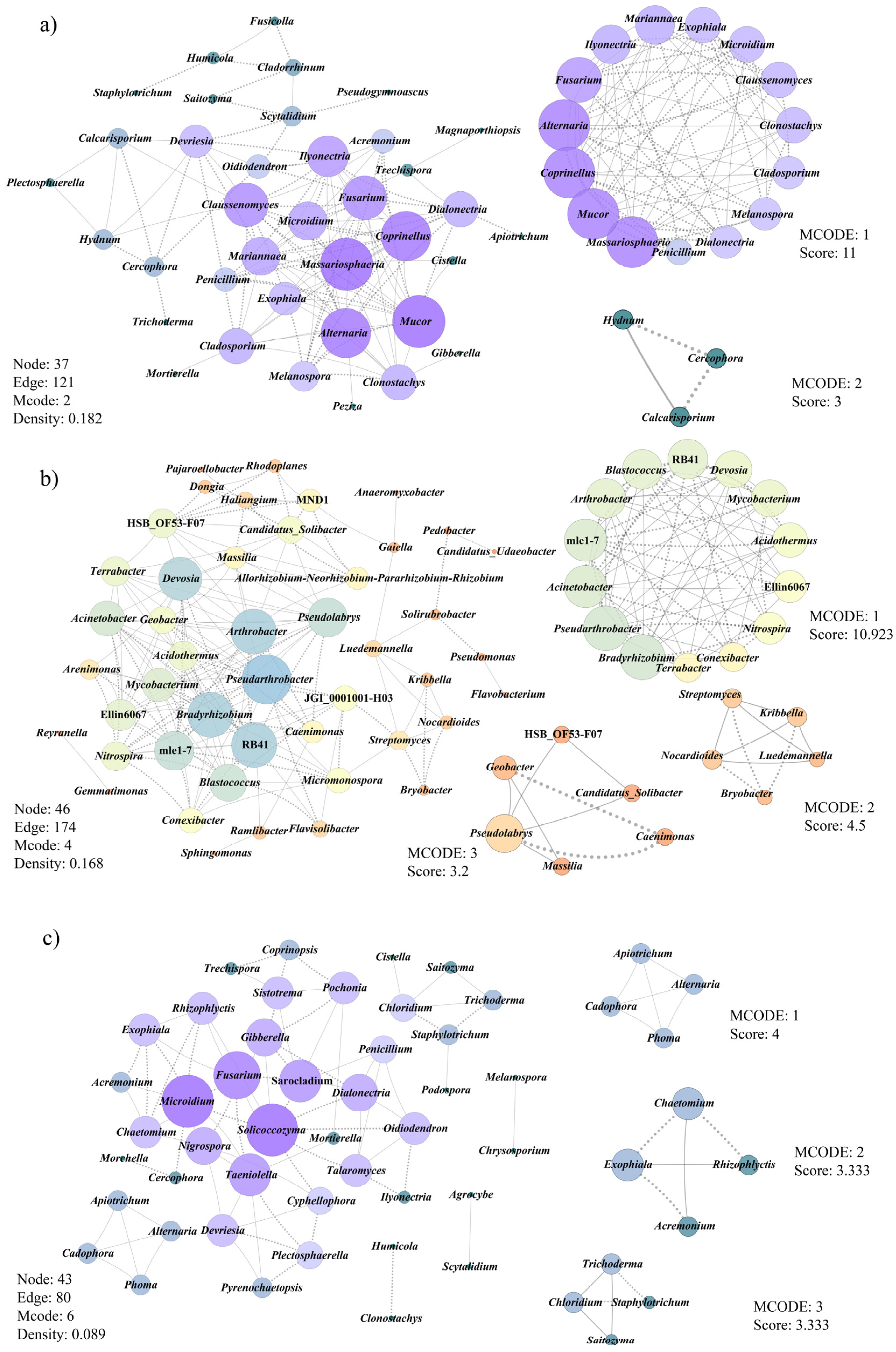
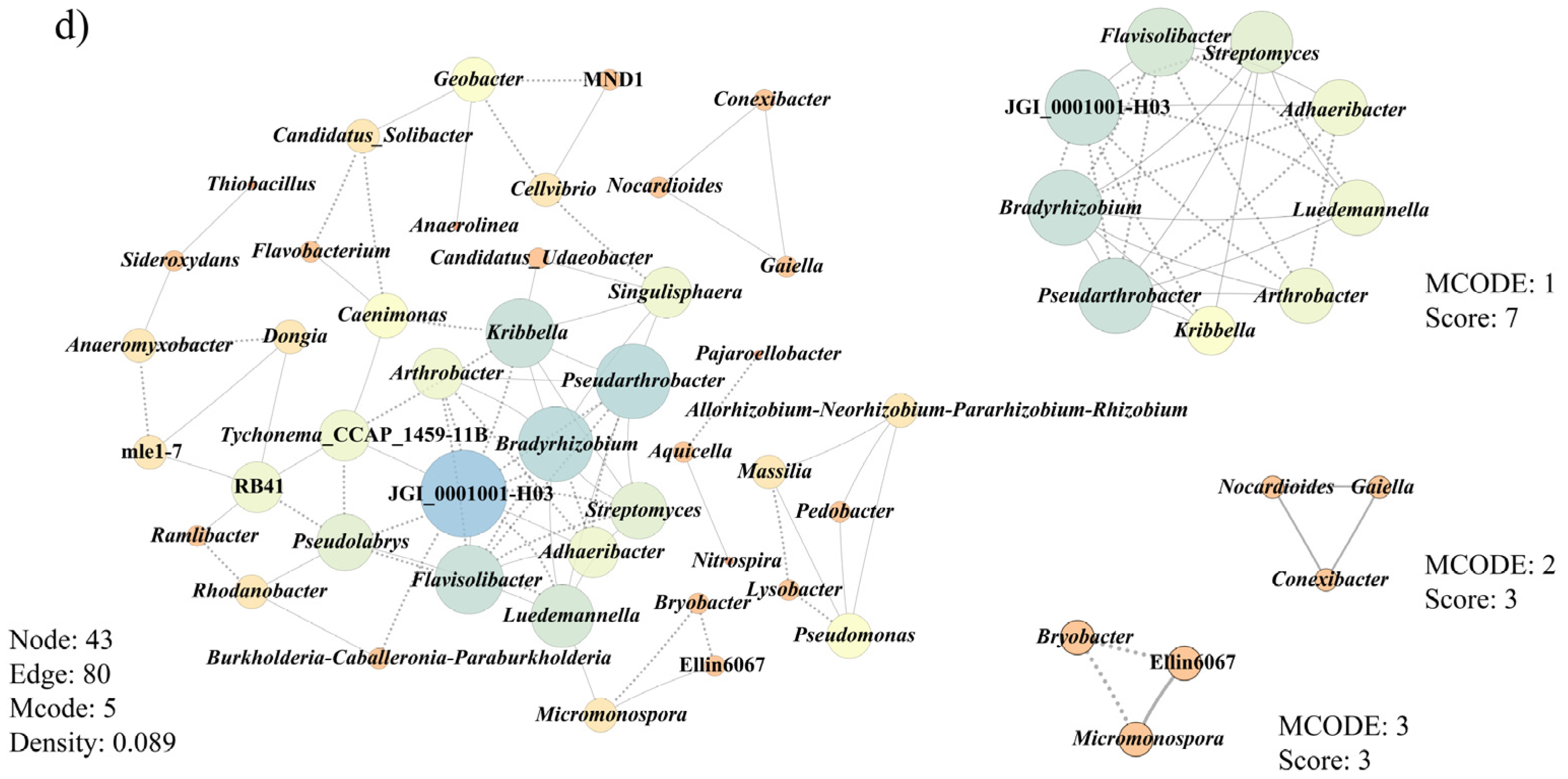
3.6. Co-Occurrence Patterns of Microbial Communities Associated with Morel Production
4. Discussion
4.1. Microbial Community Dynamics during Morel Cultivation
4.2. Positive Effects of Nitrogen Cycling on High Yields of Morels
4.3. Pathogenic Fungi Affect Morel Fructification
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Du, X.-H.; Zhao, Q.; Yang, Z.-L. A review on research advances, issues, and perspectives of morels. Mycology 2015, 6, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.-H.; Yang, Z.-L. Mating Systems in True Morels (Morchella). Microbiol. Mol. Biol. Rev. 2021, 85, e00220-20. [Google Scholar] [CrossRef] [PubMed]
- Taşkin, H.; Doǧan, H.H.; Büyükalaca, S. Morchella galilaea, an autumn species from Turkey. Mycotaxon 2015, 130, 215–221. [Google Scholar] [CrossRef]
- Matočec, N.; Kušan, I.; Mrvoš, D.; Raguzin, E. The autumnal occurrence of the vernal genus Morchella (Ascomycota, Fungi). Nat. Croat. 2014, 23, 163–177. [Google Scholar]
- Dissanayake, A.A.; Mills, G.L.; Bonito, G.; Rennick, B.; Nair, M.G. Chemical Composition and Anti-Inflammatory and Antioxidant Activities of Extracts from Cultivated Morel Mushrooms, Species of Genus Morchella (Ascomycota). Int. J. Med. Mushrooms 2021, 23, 73–83. [Google Scholar] [CrossRef]
- Wu, H.; Chen, J.; Li, J.; Liu, Y.; Park, H.J.; Yang, L. Recent advances on bioactive ingredients of Morchella esculenta. Appl. Biochem. Biotechnol. 2021, 193, 4197–4213. [Google Scholar] [CrossRef]
- Roze, M.E. Adherence de la base d’appareils ascospores de Morchella sur Helianthus tuberosus. Acta Bot. Gall. 1882, 19, 166–167. [Google Scholar]
- Ower, R. Notes on the development of the morel ascocarp: Morchella Esculenta. Mycologia 1982, 74, 142–144. [Google Scholar] [CrossRef]
- Du, X.-H.; Wang, H.; Sun, J.; Xiong, L.; Yu, J. Hybridization, characterization and transferability of SSRs in the genus Morchella. Fungal Biol. 2019, 123, 528–538. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, H.; Zhang, Y.; Dong, C. Artificial cultivation of true morels: Current state, issues and perspectives. Crit. Rev. Biotechnol. 2018, 38, 259–271. [Google Scholar] [CrossRef]
- Du, X.-H.; Wu, D.-M.; He, G.-Q.; Wei, W.; Xu, N.; Li, T.-L. Six new species and two new records of Morchella in China using phylogenetic and morphological analyses. Mycologia 2019, 111, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Lyu, M.-L.; Li, L.; Huang, W.; Zhang, Y.-F.; Hao, Z. Temptation and trap of morel industry in China. J. Fungal Res. 2021, 19, 232–237. (In Chinese) [Google Scholar]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel production related to soil microbial diversity and evenness. Microbiol. Spectr. 2021, 9, e00229-21. [Google Scholar] [CrossRef] [PubMed]
- Mer, G.S.; Dubey, R.C. Formation of sclerotial spawn, conidia and fruiting body initials in Morchella esculenta. Int. J. Environ. Biol. 1996, 1, 35–38. [Google Scholar]
- Yu, F.-M.; Zhao, Q. Genetic diversity of hyphae sprouted from different single spores and different germ pores of morels. Mycosystema 2020, 39, 1–13. (In Chinese) [Google Scholar]
- Lan, Y.-F.; Cong, Q.-Q.; Wang, Q.-W.; Tang, L.-N.; Li, X.-M.; Yu, Q.-W.; Cui, X.; An, X.-R.; Yu, C.-X.; Kong, F.-H.; et al. First report of Cladobotryum protrusum causing cobweb disease on cultivated Morchella importuna. Plant Dis. 2020, 104, 1–26. [Google Scholar] [CrossRef]
- He, P.-X.; Li, C.-C.; Cai, Y.-L.; Zhang, Y.; Bian, Y.-B.; Liu, W. First report of pileus rot disease on cultivated Morchella importuna caused by Diploöspora longispora in China. J. Gen. Plant Pathol. 2018, 84, 65–69. [Google Scholar] [CrossRef]
- He, X.-L.; Peng, W.-H.; Miao, R.-Y.; Tang, J.; Chen, Y.; Liu, L.-X.; Wang, D.; Gan, B.-C. White mold on cultivated morels caused by Paecilomyces penicillatus. FEMS Microbiol. Lett. 2017, 364, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.-P.; Chen, K.; Wang, G.-Z.; Bian, Y.-B. First report of stipe rot disease on Morchella importuna caused by Fusarium incarnatum—F. equiseti species complex in China. Plant Dis. 2016, 100, 2530. [Google Scholar] [CrossRef]
- Lohberger, A.; Spangenberg, J.E.; Ventura, Y.; Bindschedler, S.; Verrecchia, E.P.; Bshary, R.; Junier, P. Effect of organic carbon and nitrogen on the interactions of Morchella spp. and bacteria dispersing on their mycelium. Front. Microbiol. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kalyoncu, F.; Oskay, M.; Kalyoncu, M. The effects of some environmental parameters on mycelial growth of six Morchella species. J. Pure Appl. Microbiol. 2009, 3, 467–472. [Google Scholar]
- Tan, H.; Kohler, A.; Miao, R.-Y.; Liu, T.H.; Zhang, Q.; Zhang, B.; Jiang, L.; Wang, Y.; Xie, L.-Y.; Tang, J.; et al. Multi-omic analyses of exogenous nutrient bag decomposition by the black morel Morchella importuna reveal sustained carbon acquisition and transferring. Environ. Microbiol. 2019, 21, 3909–3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; He, X.-H.; Chater, C.C.C.; Perez-Moreno, J.; Yu, F.-Q. Microbiome community structure and functional gene partitioning in different micro-niches within a sporocarp-forming fungus. Front. Microbiol. 2021, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gohar, D.; Pent, M.; Poldmaa, K.; Bahram, M. Bacterial community dynamics across developmental stages of fungal fruiting bodies. FEMS Microbiol. Ecol. 2020, 96, 1–8. [Google Scholar] [CrossRef]
- Longley, R.; Benucci, G.M.N.; Mills, G.; Bonito, G. Fungal and bacterial community dynamics in substrates during the cultivation of morels (Morchella rufobrunnea) indoors. FEMS Microbiol. Lett. 2019, 366, 1–8. [Google Scholar] [CrossRef]
- Benucci, G.M.N.; Longley, R.; Zhang, P.; Zhao, Q.; Bonito, G.; Yu, F.-Q. Microbial communities associated with the black morel Morchella sextelata cultivated in greenhouses. PeerJ 2019, 2019, e7744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.-S.; Long, L.; Hu, Z.-Y.; Yu, X.-R.; Liu, Q.-Y.; Bao, J.-K.; Long, Z.-F. Analyses of artificial morel soil bacterial community structure and mineral element contents in ascocarp and the cultivated soil. Can. J. Microbiol. 2019, 65, 738–749. [Google Scholar] [CrossRef]
- McGee, C.F. Microbial ecology of the Agaricus bisporus mushroom cropping process. Appl. Microbiol. Biotechnol. 2018, 102, 1075–1083. [Google Scholar] [CrossRef]
- Cao, G.-T.; Song, T.-T.; Shen, Y.-Y.; Jin, Q.-L.; Feng, W.-L.; Fan, L.J.; Cai, W.-M. Diversity of bacterial and fungal communities in wheat straw compost for Agaricus bisporus cultivation. HortScience 2019, 54, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Ren, F.; Zhang, Y.-G.; Yu, H.; Zhang, Y.-A. Ganoderma lucidum cultivation affect microbial community structure of soil, wood segments and tree roots. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Yan, L.-J.; Li, Q.; Zou, J.; Tan, H.; Tan, W.; Peng, W.-H.; Li, X.-L.; Zhang, X.-P. Dynamic succession of substrate- associated bacterial composition and function during Ganoderma lucidum growth. PeerJ 2018, 2018, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.-H.; Bao, D.-P.; Guo, T.; Li, Y.; Ji, G.-Y.; Ji, K.-P.; Tan, Q. Bacterial profiling and dynamic succession analysis of Phlebopus portentosus casing soil using miseq sequencing. Front. Microbiol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wang, Y.-L.; Zhang, P.; Yu, F.-Q.; Perez-moreno, J. Macrofungi cultivation in shady forest areas significantly increases microbiome diversity, abundance and functional capacity in soil furrows. J. Fungi 2021, 7, 775. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, J.; Preston, G.M. Growing edible mushrooms: A conversation between bacteria and fungi. Environ. Microbiol. 2020, 22, 858–872. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, Y.-Z.; Zhang, P.-F.; Trivedi, P.; Riera, N.; Wang, Y.-Y.; Liu, X.; Fan, G.-Y.; Tang, J.-J.; Coletta-Filho, H.D.; et al. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pion, M.; Spangenberg, J.E.; Simon, A.; Bindschedler, S.; Flury, C.; Chatelain, A.; Bshary, R.; Job, D.; Junier, P. Bacterial farming by the fungus Morchella crassipes. Proc. R. Soc. B Biol. Sci. 2013, 280, 20132242. [Google Scholar] [CrossRef] [Green Version]
- Braat, N.; Koster, M.C.; Wösten, H.A. Beneficial interactions between bacteria and edible mushrooms. Fungal Biol. Rev. 2022, 39, 67–72. [Google Scholar] [CrossRef]
- Colauto, N.B.; Fermor, T.R.; Eira, A.F.; Linde, G.A. Pseudomonas putida stimulates primordia on Agaricus bitorquis. Curr. Microbiol. 2016, 72, 482–488. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Kim, J.S.; Crowley, D.E.; Cho, B.G. Growth promotion of the edible fungus Pleurotus ostreatus by fluorescent pseudomonads. FEMS Microbiol. Lett. 2003, 218, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.M.; Cho, K.M. Identification of auxin from Pseudomonas sp. P7014 for the rapid growth of Pleurotus eryngii mycelium. Korean J. Microbiol. 2014, 50, 15–21. [Google Scholar] [CrossRef]
- Orlofsky, E.; Zabari, L.; Bonito, G.; Masaphy, S. Changes in soil bacteria functional ecology associated with Morchella rufobrunnea fruiting in a natural habitat. Environ. Microbiol. 2021, 23, 6651–6662. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Correspondence QIIME allows analysis of high- throughput community sequencing data intensity normalization improves color calling in SOLiD sequencing. Nat. Publ. Gr. 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Blaxter, M.; Mann, J.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1935–1943. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The ribosomal database project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Chao, A.; Yang, M.C.K. Stopping rules and estimation for recapture debugging with unequal failure rates. Biometrika 1993, 80, 193–201. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Simpson, E. Measurment of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The Theory of Mathematical Communication; University of Illinois Press: Champaign, IL, USA, 1949. [Google Scholar]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Tan, Y.; Cui, Y.-S.; Li, H.-Y.; Kuang, A.-X.; Li, X.-R.; Wei, Y.-L.; Ji, X.-L. Rhizospheric soil and root endogenous fungal diversity and composition in response to continuous Panax notoginseng cropping practices. Microbiol. Res. 2017, 194, 10–19. [Google Scholar] [CrossRef]
- Yang, Y.-Z.; Fang, M.-K.; Wu, M.-Y.; Zhang, H.-S.; Li, H.-Z.; Zhu, J.-Q. Differences in the soil bacterial communities under organic farming and conventional farming modes revealed by 16S rDNA sequencing. J. Biobased Mater. Bioenergy 2021, 15, 10–19. [Google Scholar] [CrossRef]
- Yin, S.-J.; Suo, F.-Y.; Kong, Q.-X.; You, X.-W.; Zhang, X.; Yuan, Y.; Yu, X.-Y.; Cheng, Y.-D.; Sun, R.-X.; Zheng, H.; et al. Biochar enhanced growth and biological nitrogen fixation of wild soybean (Glycine max subsp. soja Siebold & Zucc.) in a Coastal Soil of China. Agriculture 2021, 11, 1246. [Google Scholar] [CrossRef]
- Buckley, E. Phenotypic and Genomic Insights into Ultraviolet Resistance of Arthrobacter and Pseudarthrobacter Isolated from Desert Soil. Ph.D. Thesis, Auckland University of Technology, Auckland, New Zealand, 2020. [Google Scholar]
- Barbieri, E.; Ceccaroli, P.; Saltarelli, R.; Guidi, C.; Potenza, L.; Basaglia, M.; Fontana, F.; Baldan, E.; Casella, S.; Ryahi, O.; et al. New evidence for nitrogen fixation within the Italian white truffle Tuber magnatum. Fungal Biol. 2010, 114, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Oh, S.Y.; Lee, Y.M.; Jang, Y.; Jang, S.; Kim, C.; Lim, Y.W.; Kim, J.J. Successional variation in the soil microbial community in Odaesan National Park, Korea. Sustainability 2020, 12, 4795. [Google Scholar] [CrossRef]
- Rivas, R.; Velázquez, E.; Willems, A.; Vizcaíno, N.; Subba-Rao, N.S.; Mateos, P.F.; Gillis, M.; Dazzo, F.B.; Martínez-Molina, E. A new species of Devosia that forms a unique nitrogen-fixing root-nodule symbiosis with the aquatic legume Neptunia natans (L.f.) Druce. Appl. Environ. Microbiol. 2002, 68, 5217–5222. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Zeng, L.-Y.; Luo, X.-S.; Xiong, X.; Wen, S.-L.; Wang, B.-R.; Chen, W.-L.; Huang, Q.-Y. Shifts in Nitrobacter- and Nitrospira-like nitrite-oxidizing bacterial communities under long-term fertilization practices. Soil Biol. Biochem. 2018, 124, 118–125. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.-M.; Tang, Y.-J.; Xing, Y.-M.; Qiao, P.; Li, Y.; Liu, P.-G.; Guo, S.-X. Chinese black truffle-associated bacterial communities of Tuber indicum from different geographical regions with nitrogen fixing bioactivity. Front. Microbiol. 2019, 2515. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Yu, Y.; Tang, J.; Liu, T.-T.; Miao, R.-Y.; Huang, Z.-Q.; Martin, F.M.; Peng, W.-H. Build your own mushroom soil: Microbiota succession and nutritional accumulation in semi-synthetic substratum drive the fructification of a soil-saprotrophic morel. Front. Microbiol. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Chen, C.; Fu, R.-T.; Wang, J.; Li, X.-Y.; Chen, X.-J.; Li, Q.; Lu, D.-H. Genome sequence and transcriptome profiles of pathogenic fungus Paecilomyces penicillatus reveal its interactions with edible fungus Morchella importuna. Comput. Struct. Biotecnol. J. 2021, 19, 2607–2617. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Code of Soil Sample | Detail in Morel Production | Group I | Group II | Group III | Location |
|---|---|---|---|---|---|---|
| 1 | NC | Control soil | MUCS | Songming, Yunnan | ||
| NF | high yield | MCS | H | |||
| NN | low yield | MCS | L | |||
| NSN | no yield | MCS | N | |||
| 2 | OC | Control | MUCS | Songming, Yunnan | ||
| OO | Stop cultivating morels for a year | MUSC | ||||
| OCP | high yield | MCS | H | |||
| OCF | high yield | MCS | H | |||
| ORF | high yield | MCS | H | |||
| 3 | GHC | Control | MUCS | Guanghan, Sichuan | ||
| GH1 | no yield | MCS | N | |||
| GH2 | low yield with disease | MCS | L | |||
| GH3 | no yield | MCS | N | |||
| GH4 | high yield | MCS | H | |||
| GH5 | high yield | MCS | H | |||
| 4 | SM0 | no yield | N | Songming, Yunnan | ||
| SM1 | high yield | H | ||||
| SM2 | high yield | H | ||||
| SM3 | high yield | H | ||||
| 5 | WD1 | high yield | H | Wuding, Yunnan | ||
| WD2 | high yield | H | ||||
| WD3 | high yield | H | ||||
| 6 | DY1 | no yield | N | Deyang, Sichuan |
| Factor | Fungal Community | Bacterial Community | ||||||
|---|---|---|---|---|---|---|---|---|
| Df | F. Model | R2 | p | Df | F. Model | R2 | p | |
| Soil samples | 22 | 15.414 | 0.831 | 0.001 | 22 | 7.088 | 0.693 | 0.001 |
| Residuals | 69 | 0.169 | 69 | 0.307 | ||||
| Total | 91 | 1.000 | 91 | 1.000 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, F.-M.; Jayawardena, R.S.; Thongklang, N.; Lv, M.-L.; Zhu, X.-T.; Zhao, Q. Morel Production Associated with Soil Nitrogen-Fixing and Nitrifying Microorganisms. J. Fungi 2022, 8, 299. https://doi.org/10.3390/jof8030299
Yu F-M, Jayawardena RS, Thongklang N, Lv M-L, Zhu X-T, Zhao Q. Morel Production Associated with Soil Nitrogen-Fixing and Nitrifying Microorganisms. Journal of Fungi. 2022; 8(3):299. https://doi.org/10.3390/jof8030299
Chicago/Turabian StyleYu, Feng-Ming, Ruvishika Shehali Jayawardena, Naritsada Thongklang, Meng-Lan Lv, Xue-Tai Zhu, and Qi Zhao. 2022. "Morel Production Associated with Soil Nitrogen-Fixing and Nitrifying Microorganisms" Journal of Fungi 8, no. 3: 299. https://doi.org/10.3390/jof8030299