
Polyketide Derivatives from the Endophytic Fungus Phaeosphaeria sp. LF5 Isolated from Huperzia serrata and Their Acetylcholinesterase Inhibitory Activities


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Fungal Material
2.3. Fermentation and Extraction
2.4. Isolation and Purification
2.5. Acetylcholinesterase Inhibitory Activity In Vitro Assay
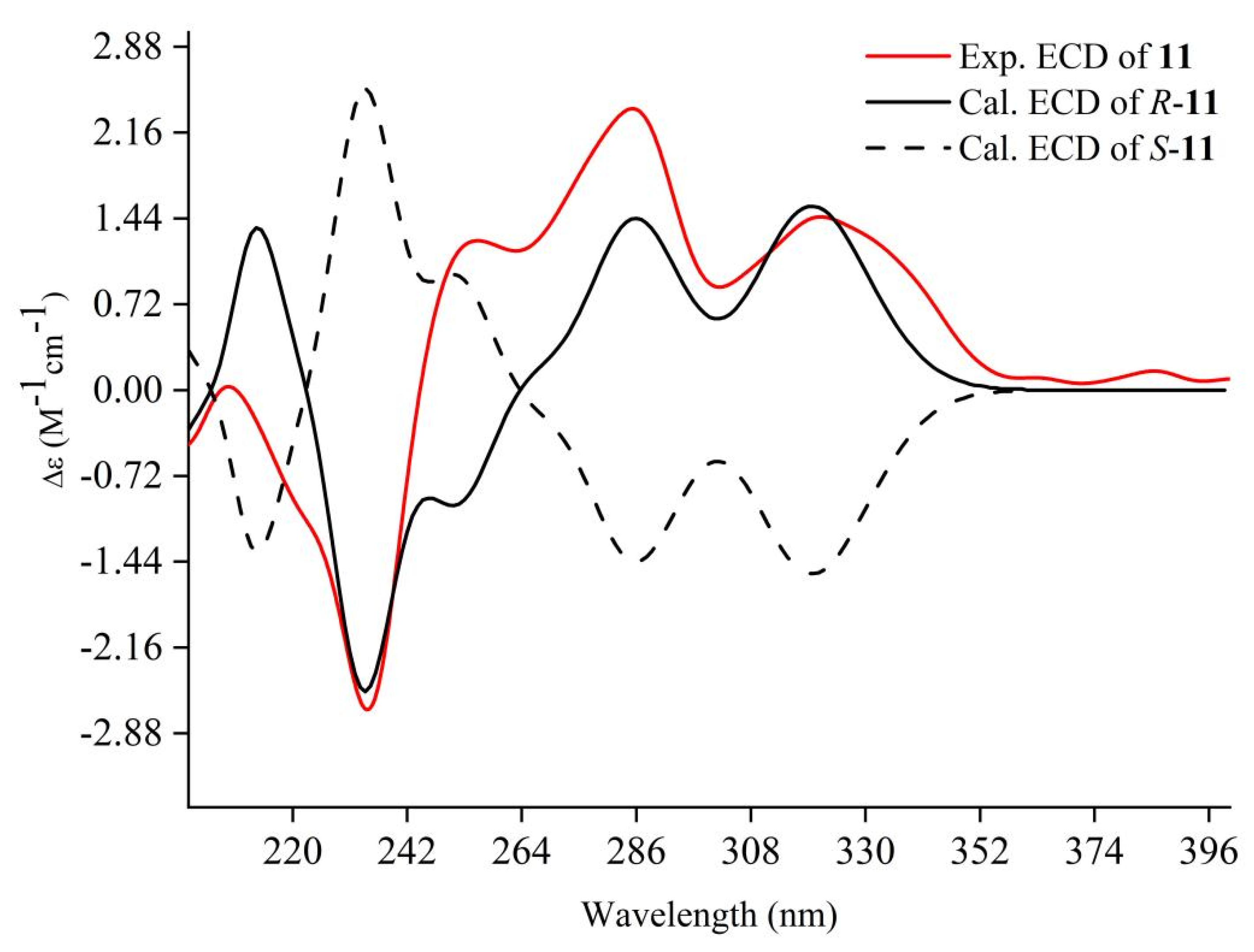
2.6. ECD Calculations
3. Results and Discussion
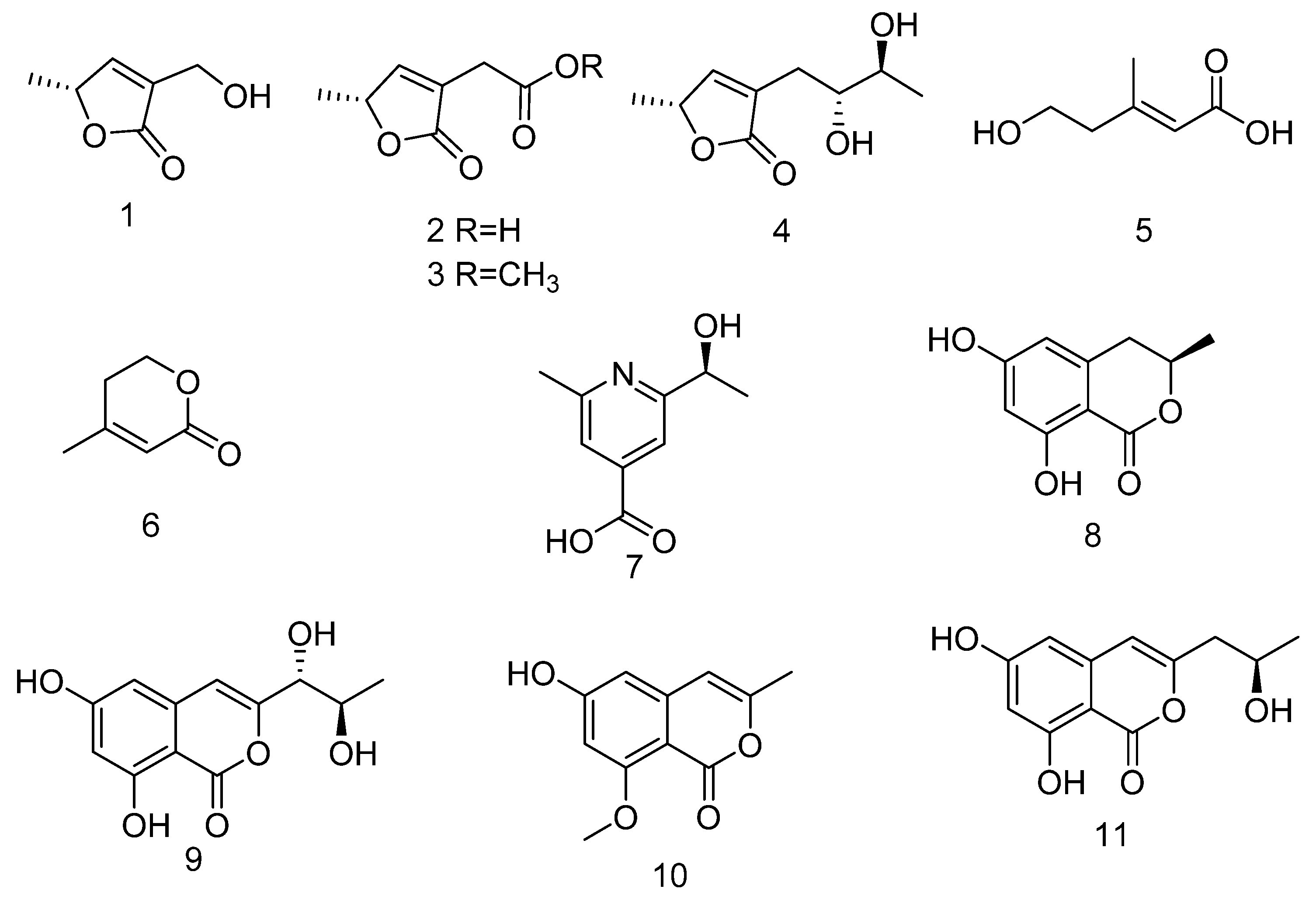
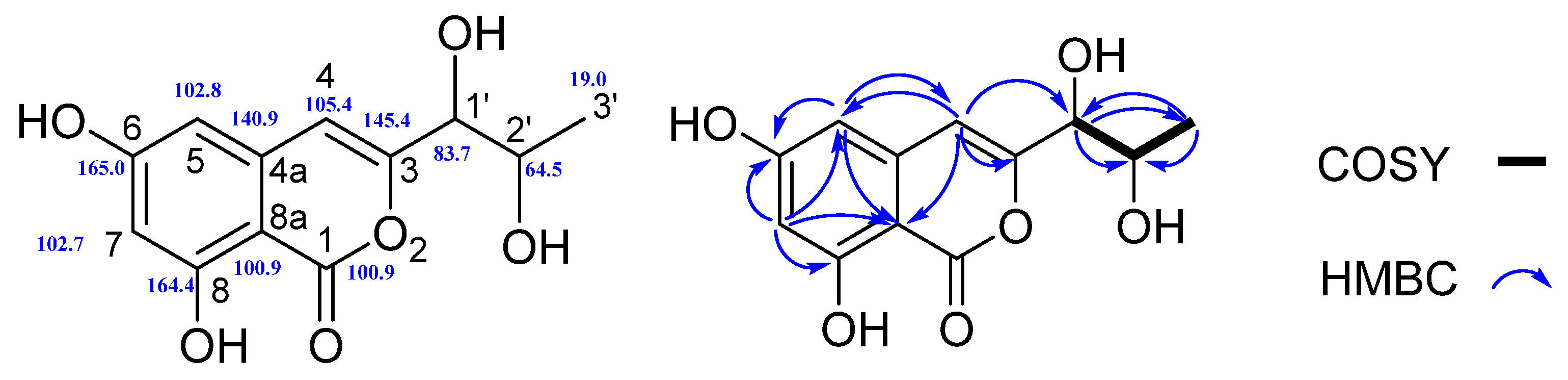
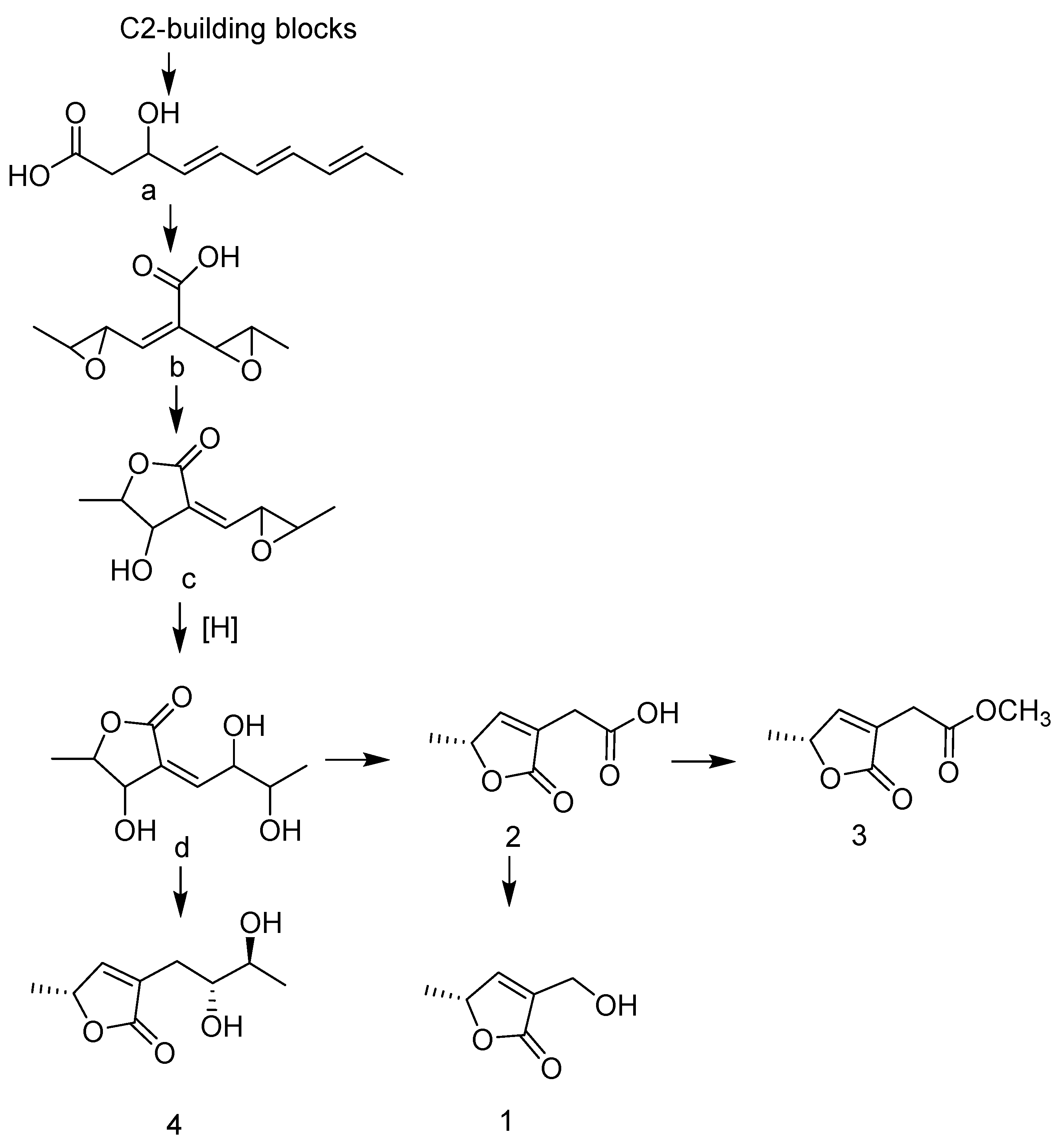
Structure Elucidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Plant endophytes and epiphytes: Burgeoning sources of known and “unknown” cytotoxic and antibiotic agents. Planta Med. 2020, 86, 13–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.B.; Zhang, G.; Li, S.M.; He, Z.H.; Yan, Q.X.; Lin, Y.K.; Xie, C.L.; Xia, J.M.; Luo, Z.H.; Luo, L.Z.; et al. Asperochratides A–J, Ten new polyketides from the deep-sea-derived Aspergillus ochraceus. Bioorg. Chem. 2020, 105, 104349. [Google Scholar] [CrossRef] [PubMed]
- Venugopalan, A.; Srivastava, S. Endophytes as in vitro production platforms of high value plant secondary metabolites. Biotechnol. Adv. 2015, 33, 873–887. [Google Scholar] [CrossRef]
- Gakuubi, M.M.; Munusamy, M.; Liang, Z.X.; Ng, S.B. Fungal endophytes: A promising frontier for discovery of novel bioactive compounds. J. Fungi 2021, 7, 786. [Google Scholar] [CrossRef]
- Deshmukh, S.K.; Gupta, M.K.; Prakash, V.; Saxena, S. Endophytic fungi: A source of potential antifungal compounds. J. Fungi 2018, 4, 77. [Google Scholar] [CrossRef] [Green Version]
- Pal, P.P.; Shaik, A.B.; Begum, A.S. Prospective leads from endophytic fungi for anti-inflammatory drug discovery. Planta Med. 2020, 86, 941–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshri, P.K.; Rai, N.; Verma, A.; Kamble, S.C.; Barik, S.; Mishra, P.; Singh, S.K.; Salvi, P.; Gautam, V. Biological potential of bioactive metabolites derived from fungal endophytes associated with medicinal plants. Mycol. Prog. 2021, 20, 577–594. [Google Scholar] [CrossRef]
- Zheng, R.; Li, S.; Zhang, X.; Zhao, C. Biological activities of some new secondary metabolites isolated from endophytic fungi: A review study. Int. J. Mol. Sci. 2021, 22, 959. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Baek, J.Y.; Kim, G.J.; Kim, J.; Kim, S.J.; Deyrup, S.T.; Choi, H.; Kang, K.S.; Shim, S.H. Azaphilones from an endophytic Penicillium sp. prevent neuronal cell death via inhibition of MAPKs and reduction of Bax/Bcl-2 ratio. J. Nat. Prod. 2021, 84, 2226–2237. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Sun, P.; Bhowmick, S.; Wei, Y.H.; Guo, B.; Wei, Y.H.; Mur, L.A.J.; Sun, Z.L. Secondary metabolites of endophytic fungi isolated from Huperzia serrata. Fitoterapia 2021, 155, 104970. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lai, Z.; Li, X.X.; Yan, R.M.; Zhang, Z.B.; Yang, H.L.; Zhu, D. Isolation, diversity and acetylcholinesterase inhibitory activity of the culturable endophytic fungi harboured in Huperzia serrata from Jinggang Mountain, China. World J. Microbiol. Biotechnol. 2016, 32, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, Q.G.; Zhang, Z.B.; Yan, R.M.; Zhu, D. Isolation and characterization of endophytic huperzine a-producing fungi from Huperzia serrata. J. Ind. Microbiol. Biotechnol. 2010, 38, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Ondeyka, J.; Harris, G.; Herath, K.; Zink, D.; Vicente, F.; Bills, G.; Collado, J.; Platas, G.; González del Val, A.; et al. Isolation, structure, and biological activity of phaeofungin, a cyclic lipodepsipeptide from a Phaeosphaeria sp. using the genome-wide Candida albicans fitness test. J. Nat. Prod. 2013, 76, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Karakoyun, Ç.; Küçüksolak, M.; Bilgi, E.; Doğan, G.; Çömlekçi, Y.E.; Bedir, E. Five new cardenolides transformed from oleandrin and nerigoside by Alternaria eureka 1e1bl1 and Phaeosphaeria sp. 1e4cs-1 and their cytotoxic activities. Phytochem. Lett. 2021, 41, 152–157. [Google Scholar] [CrossRef]
- Zhan, Z.J.; Jin, J.P.; Ying, Y.M.; Shan, W.G. Furanone derivatives from Aspergillus sp. xw-12, an endophytic fungus in Huperzia serrata. Helv. Chim. Acta 2011, 94, 1454–1458. [Google Scholar] [CrossRef]
- Husain, A.; Khan, S.A.; Iram, F.; Iqbal, M.A.; Asif, M. Insights into the chemistry and therapeutic potential of furanones: A versatile pharmacophore. Eur. J. Med. Chem. 2019, 191, 66–92. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.N.; Trinh, P.T.H.; Ivanets, E.V.G.; Smetanina, O.F.; Afiyatullov, S.S. Biologically active metabolites from the marine sediment-derived fungus Aspergillus flocculosus. Mar. Drugs 2019, 17, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Li, X.M.; Meng, L.H.; Wang, B.G. Polyketides from the marine mangrove-derived fungus Aspergillus ochraceus MA-15 and their activity against aquatic pathogenic bacteria. Phytochem. Lett. 2015, 12, 232–236. [Google Scholar] [CrossRef]
- Hu, H.C.; Li, C.Y.; Tsai, Y.H.; Yang, D.Y.; Chang, F.R. Secondary metabolites and bioactivities of Aspergillus ochraceopetaliformis isolated from Anthurium brownii. ACS Omega 2020, 5, 20991–20999. [Google Scholar] [CrossRef] [PubMed]
- Tianpanich, K.; Prachya, S.; Wiyakrutta, S.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Radical scavenging and antioxidant activities of isocoumarins and a phthalide from the endophytic fungus Colletotrichum sp. J. Nat. Prod. 2011, 74, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, X.; Chen, F.; Li, C.; Cao, F.; Luo, D. Setosphlides A-D, New Isocoumarin Derivatives from the Entomogenous Fungus Setosphaeria rostrate LGWB-10. Natur. Prod. Bioprosp. 2021, 11, 137–142. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ortiz, J.E.; Berkov, S.; Pigni, N.B.; Theoduloz, C.; Roitman, G.; Tapia, A.; Bastida, J.; Feresin, G. Wild Argentinian Amaryllidaceae, a new renewable source of the acetylcholinesterase inhibitor galanthamine and other alkaloids. Molecules 2012, 17, 13473–13482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sybyl Software; Version X 2.0; Tripos Associates Inc.: St. Louis, MO, USA, 2013.
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Stephens, P.J.; Harada, N. ECD cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 234614, 3-(Hydroxymethyl)-5-methylfuran-2(5h)-one. 2022. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/3-_Hydroxymethyl_-5-methylfuran-2_5h_-one (accessed on 24 January 2022).
- Patel, R.M.; Puranik, V.G.; Argade, N.P. Regio- and stereoselective selenium dioxide allylic oxidation of (E)-dialkyl alkylidenesuccinates to (Z)-allylic alcohols: Synthesis of natural and unnatural butenolides. Org. Biomol. Chem. 2011, 9, 6312–6322. [Google Scholar] [CrossRef] [PubMed]
- Widmer, J.; Keller-Schierlein, W. Stoffwechselprodukte von Mikroorganismen. 139. Mitteilung. Synthesen in der Sideramin-Reihe: Rhodotorulasäure und Dimerumsäure. Helv. Chim. Acta 1974, 57, 1904–1912. [Google Scholar] [CrossRef]
- Li, L.J.; Tao, M.H.; Chen, Y.C.; Zheng, C.X.; Huo, G.H.; Zhang, W.M. Secondary metabolites from the solid culture of marine fungal strain Penicillium sclerotiorum FS50. Mycosystema 2015, 34, 117–123. Available online: http://en.cnki.com.cn/Article_en/CJFDTOTAL-JWXT201501016.htm (accessed on 12 January 2022).
- de Sá, J.D.M.; Pereira, J.A.; Dethoup, T.; Cidade, H.; Sousa, M.E.; Rodrigues, I.C.; Costa, P.M.; Mistry, S.; Silva, A.M.S.; Kijjoa, A. Anthraquinones, diphenyl ethers, and their derivatives from the culture of the marine sponge-associated fungus Neosartorya spinosa KUFA 1047. Mar. Drugs 2021, 19, 457. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.X.; Zhang, B.Y.; Zhu, H.J.; Pan, L.; Cao, F. Bioactive metabolites from Talaromyces purpureogenus, an endophytic fungus from Panax notoginseng. Chem. Nat. Comp. 2020, 56, 974–976. [Google Scholar] [CrossRef]
- Hallock, Y.F.; Clardy, J.; Kenfield, D.S.; Strobel, G. De-O-methyldiaporthin, a phytotoxin from Drechslera siccans. Phytochemistry 1988, 27, 3123–3125. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Qu, H.R.; Bao, S.S.; Deng, Z.S.; Guo, Z.Y. Secondary metabolites from the endophytic fungus Xylariales sp. and their antimicrobial activity. Chem. Nat. Comp. 2020, 56, 530–532. [Google Scholar] [CrossRef]
- Chen, X.W.; Li, C.W.; Cui, C.B.; Hua, W.; Zhu, T.J.; Gu, Q.Q. Nine new and five known polyketides derived from a deep sea-sourced Aspergillus sp. 16-02-1. Mar. Drugs 2014, 12, 3116–3137. [Google Scholar] [CrossRef] [Green Version]
- Hussain, H.; Green, I.R. A patent review of two fruitful decades (1997–2016) of isocoumarin research. Expert Opin. Ther. Pat. 2017, 27, 1267–1275. [Google Scholar] [CrossRef]
- Xiang, P.; Ludwig-Radtke, L.; Yin, W.B.; Li, S.M. Isocoumarin formation by heterologous gene expression and modification by host enzymes. Org. Biomol. Chem. 2020, 18, 4946–4948. [Google Scholar] [CrossRef]
- Sonmez, F.; Zengin Kurt, B.; Gazioglu, I.; Basile, L.; Dag, A.; Cappello, V.; Ginex, T.; Kucukislamoglu, M.; Guccione, S. Design, synthesis and docking study of novel coumarin ligands as potential selective acetylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 32, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.; Guo, Y.H.; Li, G.; Wu, C.S.; Li, M. Metabolites of endophytic fungus Elaphocordyceps sp. Microbiol. China 2014, 41, 857–861. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1H NMR | 13C NMR | HMBC | HSQC |
|---|---|---|---|---|
| 2 | ― | 176.5 (s) | H-4, H-7 | ― |
| 3 | ― | 131.7 (s) | H-4, H-7, H-8 | ― |
| 4 | 7.36 (1H, br s) | 154.2 (d) | H-5, H-6, H-7 | 7.36 |
| 5 | 5.09 (1H, br q, 6.8) | 79.8 (d) | H-4, H-6 | 5.09 |
| 6 | 1.39 (3H, d, 6.8) | 19.1 (q) | H-5 | 1.39 |
| 7 | 2.30 (1H, br dd, 15.2, 9.1); 2.59 (1H, br d, 15.2) | 29.6 (t) | H-8, H-9 | 2.30, 2.59 |
| 8 | 3.60 (1H, m) | 74.8 (d) | H-7, H-9, H-10 | 3.60 |
| 9 | 3.58 (1H, m) | 71.6 (d) | H-8, H-10 | 3.58 |
| 10 | 1.20 (3H, d, 6.1) | 18.9 (q) | H-8, H-9 | 1.20 |
| No. | 1H NMR | 13C NMR | HMBC | HSQC(δ) |
|---|---|---|---|---|
| 2 | ― | 166.9 (s) | H-3, H-8 | ― |
| 3 | 7.89 (1H, br s) | 117.5 (d) | H-5 | 7.89 |
| 4 | ― | 142.2 (s) | ― | ― |
| 5 | 7.67 (1H, br s) | 122.6 (d) | H-3, H-10 | 7.67 |
| 6 | ― | 159.5 (s) | H-10, H-5 | ― |
| 7 | 4.89 (1H, overlapped) | 71.1 (d) | H-8 | 4.89 |
| 8 | 1.46 (3H, d, 6.6) | 24.4 (q) | H-7 | 1.36 |
| 9 | ― | 168.4 (s) | H-3, H-5 | ― |
| 10 | 2.59 (3H, s) | 23.7 (q) | H-5 | 2.59 |
| No. | 1HNMR | 13C NMR | HMBC | HSQC |
|---|---|---|---|---|
| 1 | ― | 165.54 | ― | ― |
| 3 | ― | 157.6 | H-4 | ― |
| 4 | 6.61 (1H, s) | 104.4 | ― | 6.61 |
| 4a | ― | 139.2 | ― | ― |
| 5 | 6.43 (1H, s) | 102.9 | H-4, H-7 | 6.43 |
| 6 | 10.88 (OH, s) | 165.3 | H-7 | ― |
| 7 | 6.33 (1H, s) | 101.5 | ― | 6.33 |
| 8 | 11.00 (OH, s) | 162.5 | H-7 | ― |
| 8a | ― | 97.8 | H-4, H-5, H-7 | ― |
| 1′ | 3.97 (1H, dd) | 74.7 | H-4, H-3′ | 3.97 |
| 2′ | 3.80 (1H, dq) | 67.4 | H-1′, H-3′ | 3.80 |
| 3′ | 1.12 (3H, d) | 19.7 | H-1′ | 1.12 |
| Compound | IC50 (µM) a | Compounds | IC50 (µM) a |
|---|---|---|---|
| 1 | >100 | 6 | >100 |
| 2 | >100 | 7 | >100 |
| 3 | >100 | 8 | >100 |
| 4 | 6.26 ± 0.15 | 9 | >100 |
| 5 | >100 | 10 | >100 |
| Rivastigmine | 1.82 ± 0.13 | 11 | 21.18 ± 1.53 |
| Hup-A | 0.045 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Y.; Liang, W.; Zhang, Z.; Wang, Y.; Zhang, S.; Liu, J.; Chang, J.; Ji, C.; Zhu, D. Polyketide Derivatives from the Endophytic Fungus Phaeosphaeria sp. LF5 Isolated from Huperzia serrata and Their Acetylcholinesterase Inhibitory Activities. J. Fungi 2022, 8, 232. https://doi.org/10.3390/jof8030232
Xiao Y, Liang W, Zhang Z, Wang Y, Zhang S, Liu J, Chang J, Ji C, Zhu D. Polyketide Derivatives from the Endophytic Fungus Phaeosphaeria sp. LF5 Isolated from Huperzia serrata and Their Acetylcholinesterase Inhibitory Activities. Journal of Fungi. 2022; 8(3):232. https://doi.org/10.3390/jof8030232
Chicago/Turabian StyleXiao, Yiwen, Weizhong Liang, Zhibin Zhang, Ya Wang, Shanshan Zhang, Jiantao Liu, Jun Chang, Changjiu Ji, and Du Zhu. 2022. "Polyketide Derivatives from the Endophytic Fungus Phaeosphaeria sp. LF5 Isolated from Huperzia serrata and Their Acetylcholinesterase Inhibitory Activities" Journal of Fungi 8, no. 3: 232. https://doi.org/10.3390/jof8030232