
A Dual-Plasmid-Based CRISPR/Cas9-Mediated Strategy Enables Targeted Editing of pH Regulatory Gene pacC in a Clinical Isolate of Trichophyton rubrum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Strains, Plasmids, and Growth Conditions
2.2. sgRNA Plasmid Construction
2.3. DNA Cleavage by In Vitro Assembled Cas9–RNP Complex
2.4. Protoplast Generation, Transformation of T. rubrum, and Mutant Screening
2.5. Off-Target Analysis
2.6. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
2.7. Phenotypic Characterization of pacC Mutants
3. Results
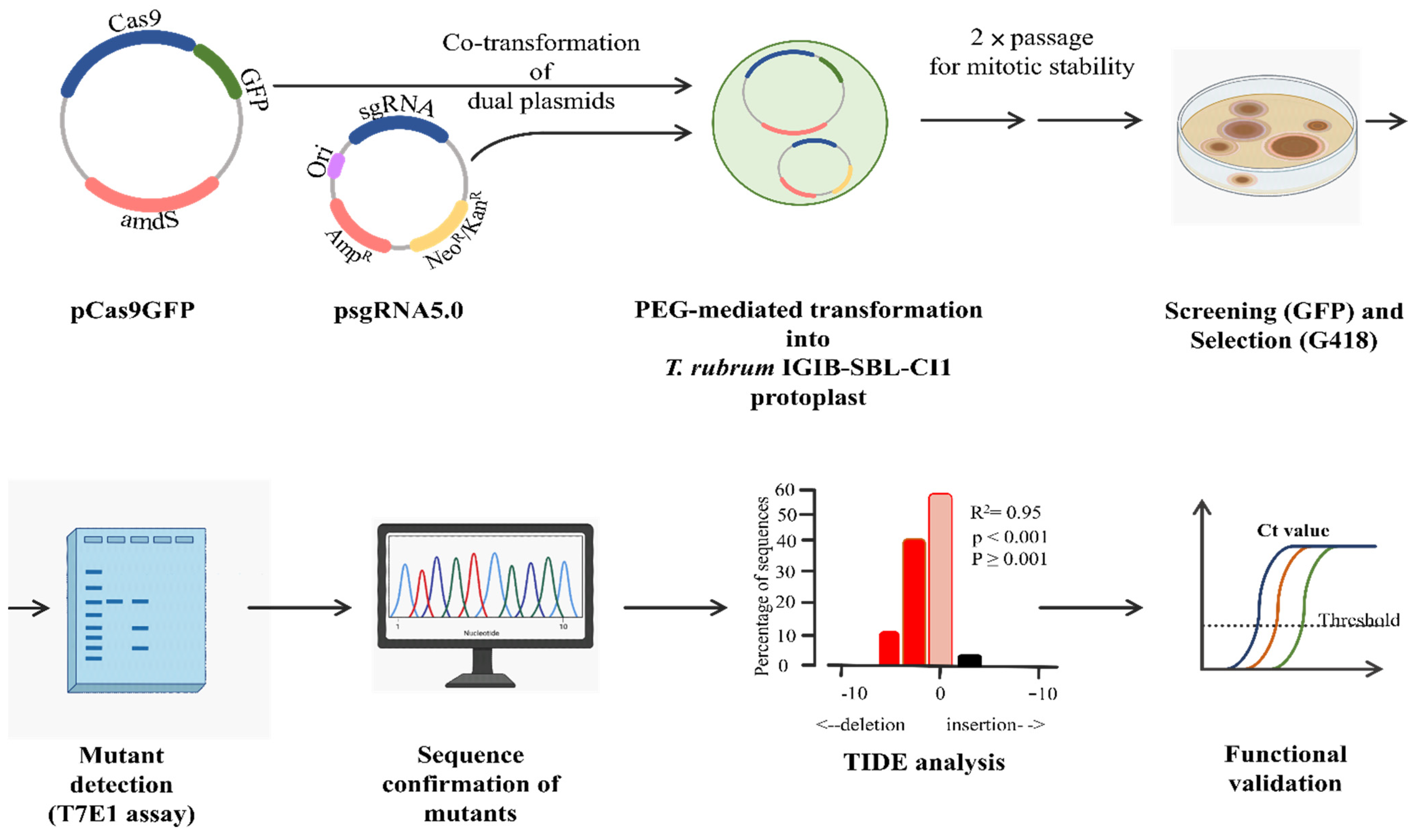
3.1. Overall Strategy
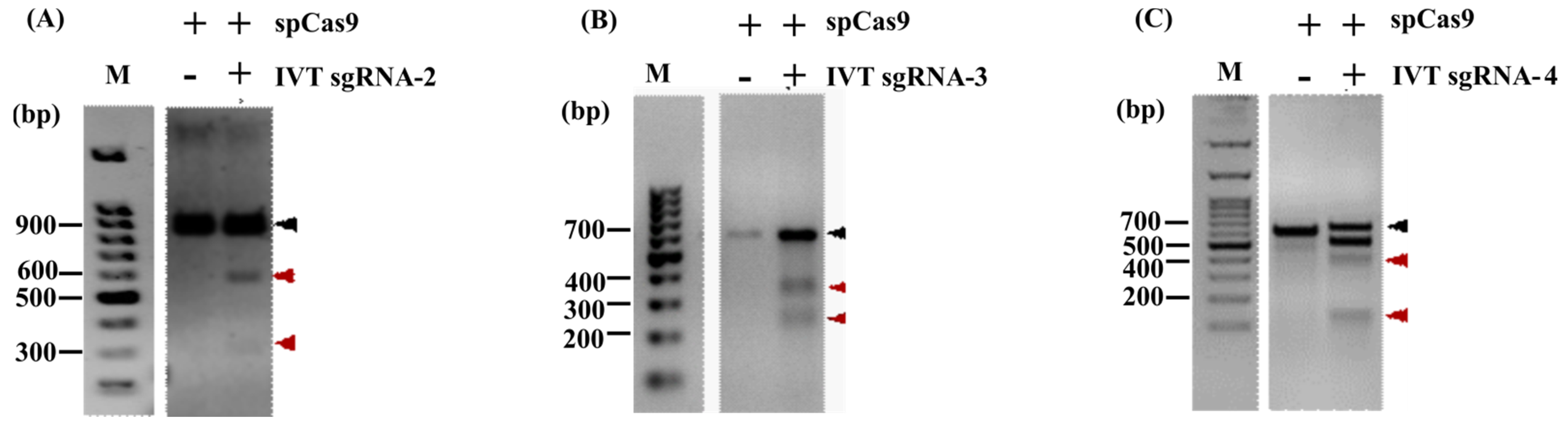
3.2. In Vitro Cleavage Assay (IVC)
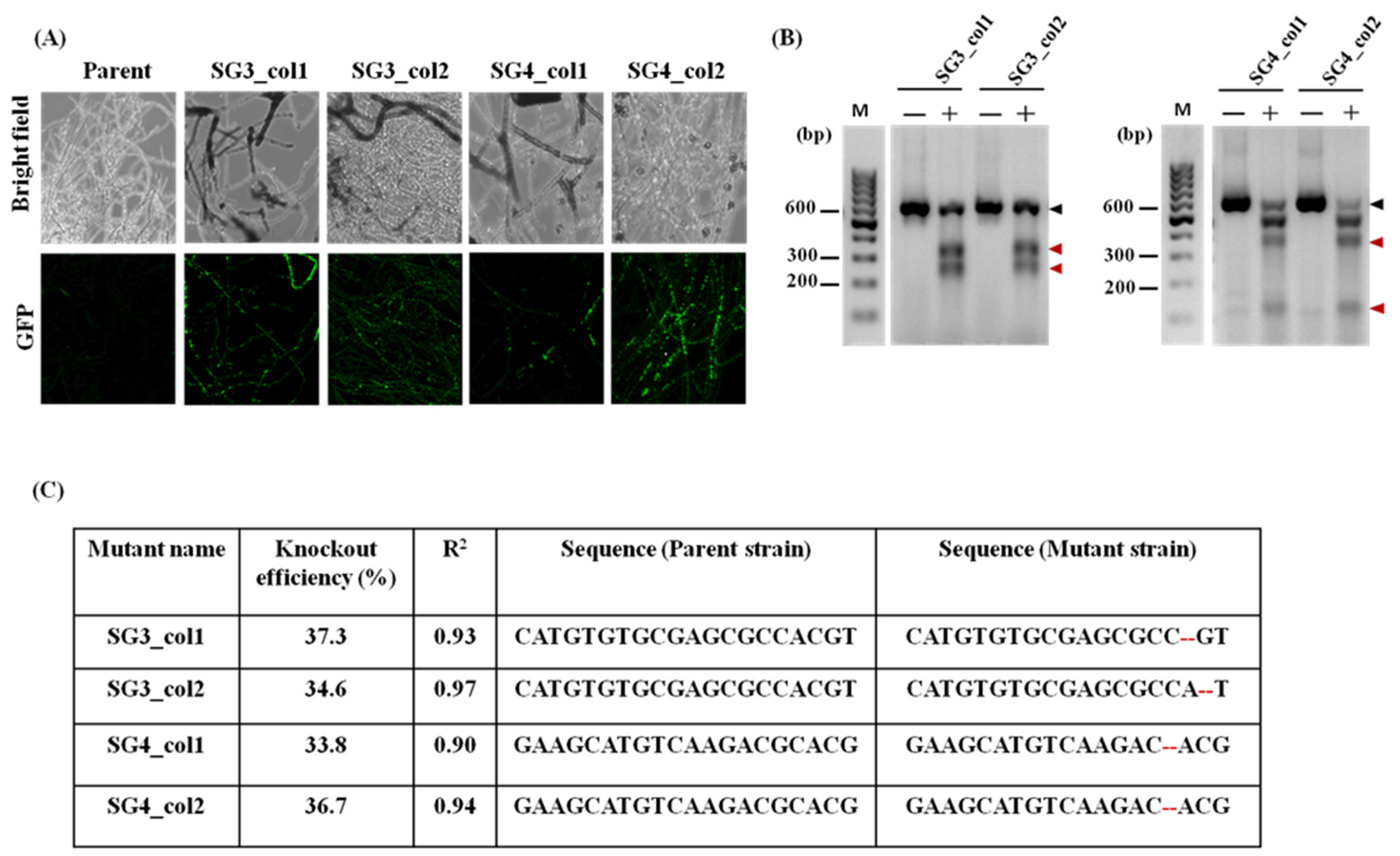
3.3. Dual-Plasmid-Based CRISPR/Cas9 Strategy Successfully Generates pacC Mutants of T. rubrum IGIB-SBL-CI1
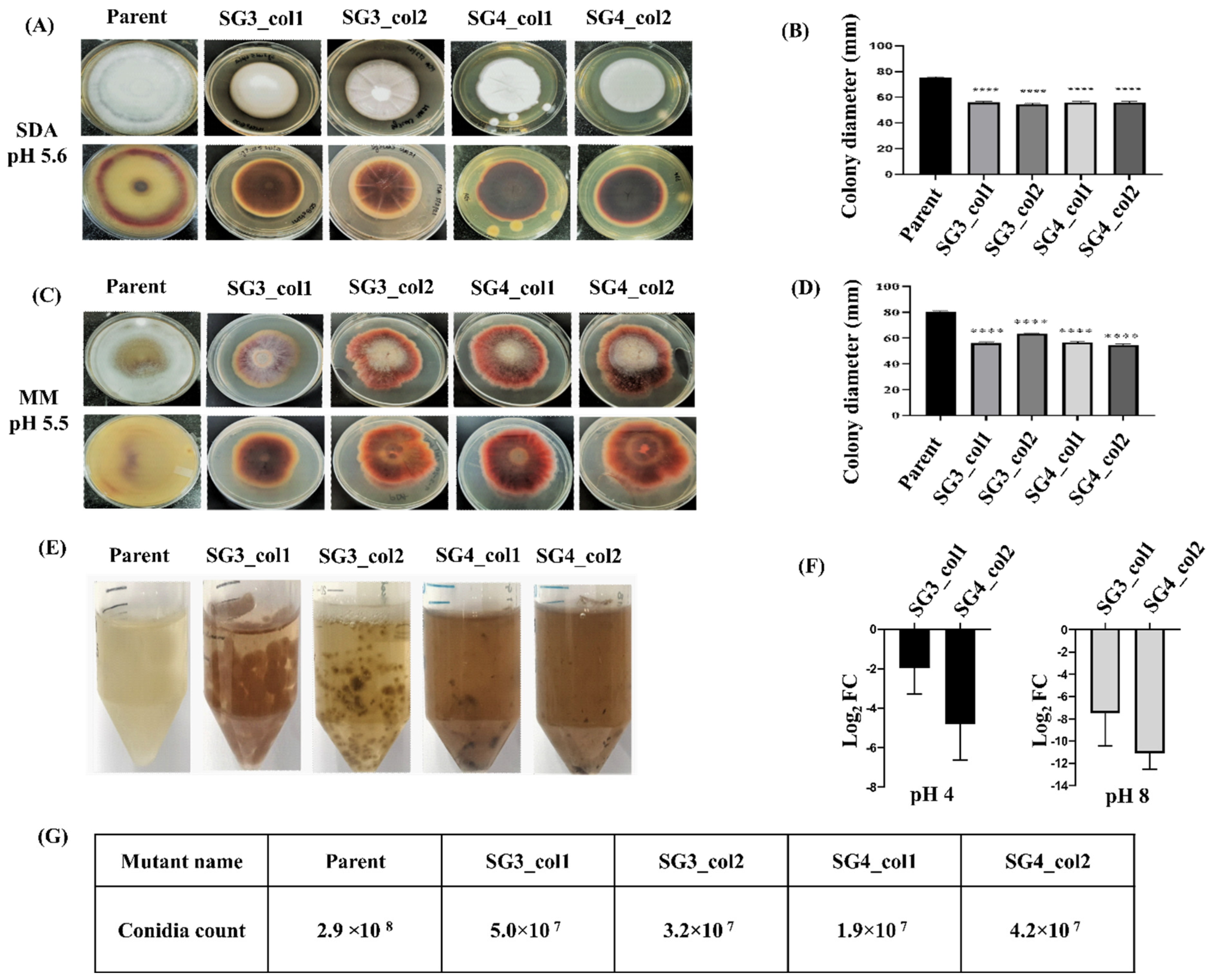
3.4. Characterization of the pacC Mutants
3.5. In Vivo Roles: Sensitivity of pacC Mutants to pH Variation and Saline Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Achterman, R.R.; White, T.C. A Foot in the Door for Dermatophyte Research. PLoS Pathog. 2012, 8, e1002564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, V.L.; Beccacece, I.; Guasconi, L.; Mena, C.J.; Cervi, L.; Chiapello, L.S. Skin Immunity to Dermatophytes: From Experimental Infection Models to Human Disease. Front. Immunol. 2020, 11, 605644. [Google Scholar] [CrossRef] [PubMed]
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Nenoff, P.; Krüger, C.; Schaller, J.; Ginter-Hanselmayer, G.; Schulte-Beerbühl, R.; Tietz, H.-J. Mycology-an update Part 2: Dermatomycoses: Clinical picture and diagnostics: CME-Article. JDDG J. Dtsch. Dermatol. Ges. 2014, 12, 749–777. [Google Scholar] [CrossRef]
- Lee, W.J.; Kim, S.L.; Jang, Y.H.; Lee, S.-J.; Kim, D.W.; Bang, Y.J.; Jun, J.B. Increasing Prevalence of Trichophyton rubrum Identified through an Analysis of 115,846 Cases over the Last 37 Years. J. Korean Med. Sci. 2015, 30, 639. [Google Scholar] [CrossRef] [Green Version]
- Zhan, P.; Liu, W. The Changing Face of Dermatophytic Infections Worldwide. Mycopathologia 2017, 182, 77–86. [Google Scholar] [CrossRef]
- Petrucelli, M.F.; de Abreu, M.H.; Cantelli, B.A.M.; Segura, G.G.; Nishimura, F.G.; Bitencourt, T.A.; Marins, M.; Fachin, A.L. Epidemiology and Diagnostic Perspectives of Dermatophytoses. J. Fungi 2020, 6, 310. [Google Scholar] [CrossRef]
- Yamada, T.; Maeda, M.; Alshahni, M.M.; Tanaka, R.; Yaguchi, T.; Bontems, O.; Salamin, K.; Fratti, M.; Monod, M. Terbinafine Resistance of Trichophyton Clinical Isolates Caused by Specific Point Mutations in the Squalene Epoxidase Gene. Antimicrob. Agents Chemother. 2017, 61, e00115-17. [Google Scholar] [CrossRef] [Green Version]
- Rudramurthy, S.M.; Shankarnarayan, S.A.; Dogra, S.; Shaw, D.; Mushtaq, K.; Paul, R.A.; Narang, T.; Chakrabarti, A. Mutation in the Squalene Epoxidase Gene of Trichophyton interdigitale and Trichophyton rubrum Associated with Allylamine Resistance. Antimicrob. Agents Chemother. 2018, 62, e02522-17. [Google Scholar] [CrossRef] [Green Version]
- Ebert, A.; Monod, M.; Salamin, K.; Burmester, A.; Uhrlaß, S.; Wiegand, C.; Hipler, U.; Krüger, C.; Koch, D.; Wittig, F.; et al. Alarming India-wide phenomenon of antifungal resistance in dermatophytes: A multicentre study. Mycoses 2020, 63, 717–728. [Google Scholar] [CrossRef]
- Jiang, Y.; Luo, W.; Verweij, P.E.; Song, Y.; Zhang, B.; Shang, Z.; Al-Hatmi, A.M.S.; Ahmed, S.A.; Wan, Z.; Li, R.; et al. Regional Differences in Antifungal Susceptibility of the Prevalent Dermatophyte Trichophyton rubrum. Mycopathologia 2021, 186, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Venkataraman, M. Antifungal resistance in superficial mycoses. J. Dermatol. Treat. 2022, 33, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Makimura, K.; Hisajima, T.; Ito, M.; Umeda, Y.; Abe, S. Genetic transformation of the dermatophyte, Trichophyton mentagrophytes, based on the use of G418 resistance as a dominant selectable marker. J. Dermatol. Sci. 2008, 49, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Grumbt, M.; Monod, M.; Staib, P. Genetic advances in dermatophytes. FEMS Microbiol. Lett. 2011, 320, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshahni, M.M.; Yamada, T. Genetic Manipulations in Dermatophytes. Mycopathologia 2017, 182, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Makimura, K.; Uchida, K.; Yamaguchi, H. Reproducible genetic transformation system for two dermatophytes, Microsporum canis and Trichophyton mentagrophytes. Med. Mycol. 2005, 43, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Makimura, K.; Abe, S. Isolation, characterization, and disruption of dnr1, the areA/nit-2-like nitrogen regulatory gene of the zoophilic dermatophyte, Microsporum canis. Med. Mycol. 2006, 44, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Fachin, A.L.; Ferreira-Nozawa, M.S.; Maccheroni, W.; Martinez-Rossi, N.M. Role of the ABC transporter TruMDR2 in terbinafine, 4-nitroquinoline N-oxide and ethidium bromide susceptibility in Trichophyton rubrum. J. Med. Microbiol. 2006, 55, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Nozawa, M.S.; Silveira, H.C.S.; Ono, C.J.; Fachin, A.L.; Rossi, A.; Martinez-Rossi, N.M. The pH signaling transcription factor PacC mediates the growth of Trichophyton rubrum on human nail in vitro. Med. Mycol. 2006, 44, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Vermout, S.; Tabart, J.; Baldo, A.; Monod, M.; Losson, B.; Mignon, B. RNA silencing in the dermatophyte Microsporum canis. FEMS Microbiol. Lett. 2007, 275, 38–45. [Google Scholar] [CrossRef]
- Yamada, T.; Makimura, K.; Hisajima, T.; Ishihara, Y.; Umeda, Y.; Abe, S. Enhanced gene replacements in Ku80 disruption mutants of the dermatophyte, Trichophyton mentagrophytes. FEMS Microbiol. Lett. 2009, 298, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Blechert, O.; Mei, H.; Zang, X.; Zheng, H.; Liang, G.; Liu, W. Auxotrophic mutations of Trichophyton rubrum created by in vitro synthesized Cas9 ribonucleoprotein. BMC Biotechnol. 2020, 20, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peres, N.T.; Sanches, P.R.; Falcão, J.P.; Silveira, H.C.; Paião, F.G.; Maranhão, F.C.; Gras, D.E.; Segato, F.; Cazzaniga, R.A.; Mazucato, M.; et al. Transcriptional profiling reveals the expression of novel genes in response to various stimuli in the human dermatophyte Trichophyton rubrum. BMC Microbiol. 2010, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, N.S.; Trevisan, G.L.; Silva Cruz, A.H.; Santos, R.S.; Peres, N.T.A.; Martinez-Rossi, N.M.; Rossi, A. Transcription of N- and O-linked mannosyltransferase genes is modulated by the pacC gene in the human dermatophyte Trichophyton rubrum. FEBS Open Biol. 2012, 2, 294–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Zheng, P.; Sun, J.; Kun, Z.; Ma, Y. Heterologous and endogenous U6 snRNA promoters enable CRISPR/Cas9 mediated genome editing in Aspergillus niger. Fungal Biol. Biotechnol. 2018, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Latka, C.; Dey, S.S.; Mahajan, S.; Prabu, R.; Jangir, P.K.; Gupta, C.; Das, S.; Ramachandran, V.G.; Bhattacharya, S.N.; Pandey, R.; et al. Genome sequence of a clinical isolate of dermatophyte, Trichophyton rubrum from India. FEMS Microbiol. Lett. 2015, 362, fnv039. [Google Scholar] [CrossRef] [Green Version]
- Gaurav, V.; Bhattacharya, S.N.; Sharma, N.; Datt, S.; Kumar, P.; Rai, G.; Singh, P.K.; Taneja, B.; Das, S. Terbinafine resistance in dermatophytes: Time to revisit alternate antifungal therapy. J. Med. Mycol. 2021, 31, 101087. [Google Scholar] [CrossRef]
- Kumar, P.; Das, S.; Tigga, R.; Pandey, R.; Bhattacharya, S.N.; Taneja, B. Whole genome sequences of two Trichophyton indotineae clinical isolates from India emerging as threats during therapeutic treatment of dermatophytosis. 3 Biotech 2021, 11, 402. [Google Scholar] [CrossRef]
- Yue, X.; Li, Q.; Wang, H.; Sun, Y.; Wang, A.; Zhang, Q.; Zhang, C. An ultrastructural study of Trichophyton rubrum induced onychomycosis. BMC Infect. Dis. 2015, 15, 532. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zheng, P.; Zhang, K.; Cairns, T.C.; Meyer, V.; Sun, J.; Ma, Y. 5S rRNA Promoter for Guide RNA Expression Enabled Highly Efficient CRISPR/Cas9 Genome Editing in Aspergillus niger. ACS Synth. Biol. 2019, 8, 1568–1574. [Google Scholar] [CrossRef]
- Jao, L.-E.; Wente, S.R.; Chen, W. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc. Natl. Acad. Sci. USA 2013, 110, 13904–13909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, S.E.; Hu, Y.; Ewen-Campen, B.; Housden, B.E.; Viswanatha, R.; Perrimon, N. CRISPR guide RNA design for research applications. FEBS J. 2016, 283, 3232–3238. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, D.; Fujihara, Y.; Satouh, Y.; Miyata, H.; Isotani, A.; Ikawa, M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci. Rep. 2013, 3, 3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, S.; Mishra, A.; Paul, D.; Ansari, A.H.; Azhar, M.; Kumar, M.; Rauthan, R.; Sharma, N.; Aich, M.; Sinha, D.; et al. Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc. Natl. Acad. Sci. USA 2019, 116, 20959–20968. [Google Scholar] [CrossRef] [Green Version]
- Meyer, V.; Ram, A.F.J.; Punt, P.J. Genetics, Genetic Manipulation, and Approaches to Strain Improvement of Filamentous Fungi. In Manual of Industrial Microbiology and Biotechnology; Baltz, R.H., Davies, J.E., Demain, A.L., Bull, A.T., Junker, B., Katz, L., Lynd, L.R., Masurekar, P., Reeves, C.D., Zhao, H., Eds.; ASM Press: Washington, DC, USA, 2014; pp. 318–329. [Google Scholar] [CrossRef]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 888. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, L.G.; Martins, M.P.; Sanches, P.R.; de Aguiar Peres, N.T.; Martinez-Rossi, N.M.; Rossi, A. Saline stress affects the pH-dependent regulation of the transcription factor PacC in the dermatophyte Trichophyton interdigitale. Braz. J. Microbiol. 2020, 51, 1585–1591. [Google Scholar] [CrossRef]
- Fincham, J.R. Transformation in fungi. Microbiol. Rev. 1989, 53, 148–170. [Google Scholar] [CrossRef]
- Nødvig, C.S.; Nielsen, J.B.; Kogle, M.E.; Mortensen, U.H. A CRISPR-Cas9 System for Genetic Engineering of Filamentous Fungi. PLoS ONE 2015, 10, e0133085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumbt, M.; Defaweux, V.; Mignon, B.; Monod, M.; Burmester, A.; Wöstemeyer, J.; Staib, P. Targeted Gene Deletion and In Vivo Analysis of Putative Virulence Gene Function in the Pathogenic Dermatophyte Arthroderma benhamiae. Eukaryot. Cell 2011, 10, 842–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva Santos, R.; Orlandelli, R.C.; Pamphile, J.A. The role of PacC and Nuc-1 transcription factors in the microbial metabolic engineering. Braz. J. Biol. Sci. 2018, 5, 213–223. [Google Scholar] [CrossRef]
- Smith, D.F.Q.; Casadevall, A. The Role of Melanin in Fungal Pathogenesis for Animal Hosts. Curr. Top. Microbiol. Immunol. 2019, 422, 1–30. [Google Scholar] [CrossRef]
- Eisenman, H.C.; Greer, E.M.; McGrail, C.W. The role of melanins in melanotic fungi for pathogenesis and environmental survival. Appl. Microbiol. Biotechnol. 2020, 104, 4247–4257. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, S.S.; Ramalingam, S.; Taneja, B. A Dual-Plasmid-Based CRISPR/Cas9-Mediated Strategy Enables Targeted Editing of pH Regulatory Gene pacC in a Clinical Isolate of Trichophyton rubrum. J. Fungi 2022, 8, 1241. https://doi.org/10.3390/jof8121241
Dey SS, Ramalingam S, Taneja B. A Dual-Plasmid-Based CRISPR/Cas9-Mediated Strategy Enables Targeted Editing of pH Regulatory Gene pacC in a Clinical Isolate of Trichophyton rubrum. Journal of Fungi. 2022; 8(12):1241. https://doi.org/10.3390/jof8121241
Chicago/Turabian StyleDey, Sanchita Sanchaya, Sivaprakash Ramalingam, and Bhupesh Taneja. 2022. "A Dual-Plasmid-Based CRISPR/Cas9-Mediated Strategy Enables Targeted Editing of pH Regulatory Gene pacC in a Clinical Isolate of Trichophyton rubrum" Journal of Fungi 8, no. 12: 1241. https://doi.org/10.3390/jof8121241