
Cryptic Species Diversity and Phylogenetic Relationship in the Rust Genus Chrysomyxa from China


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collections
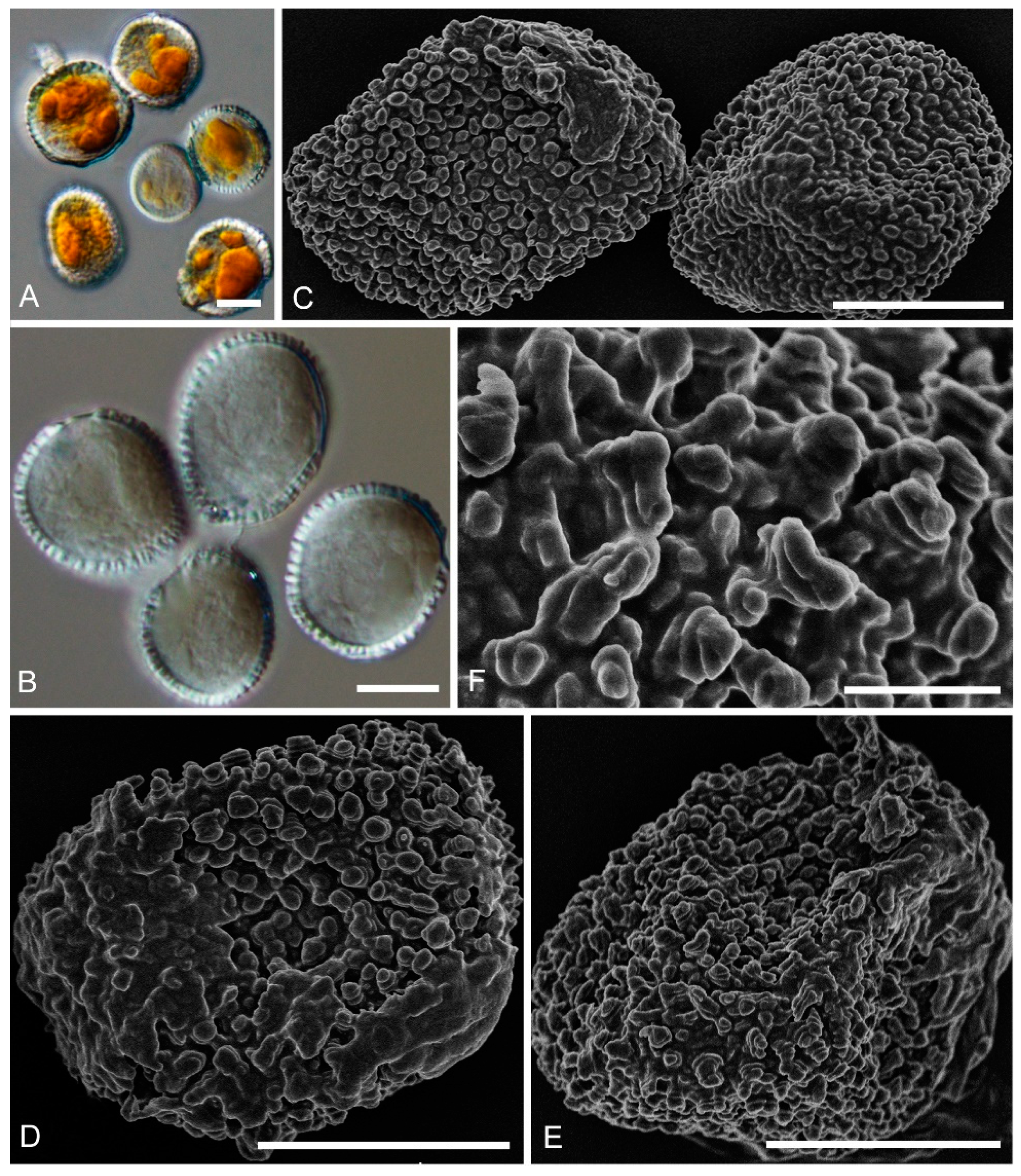
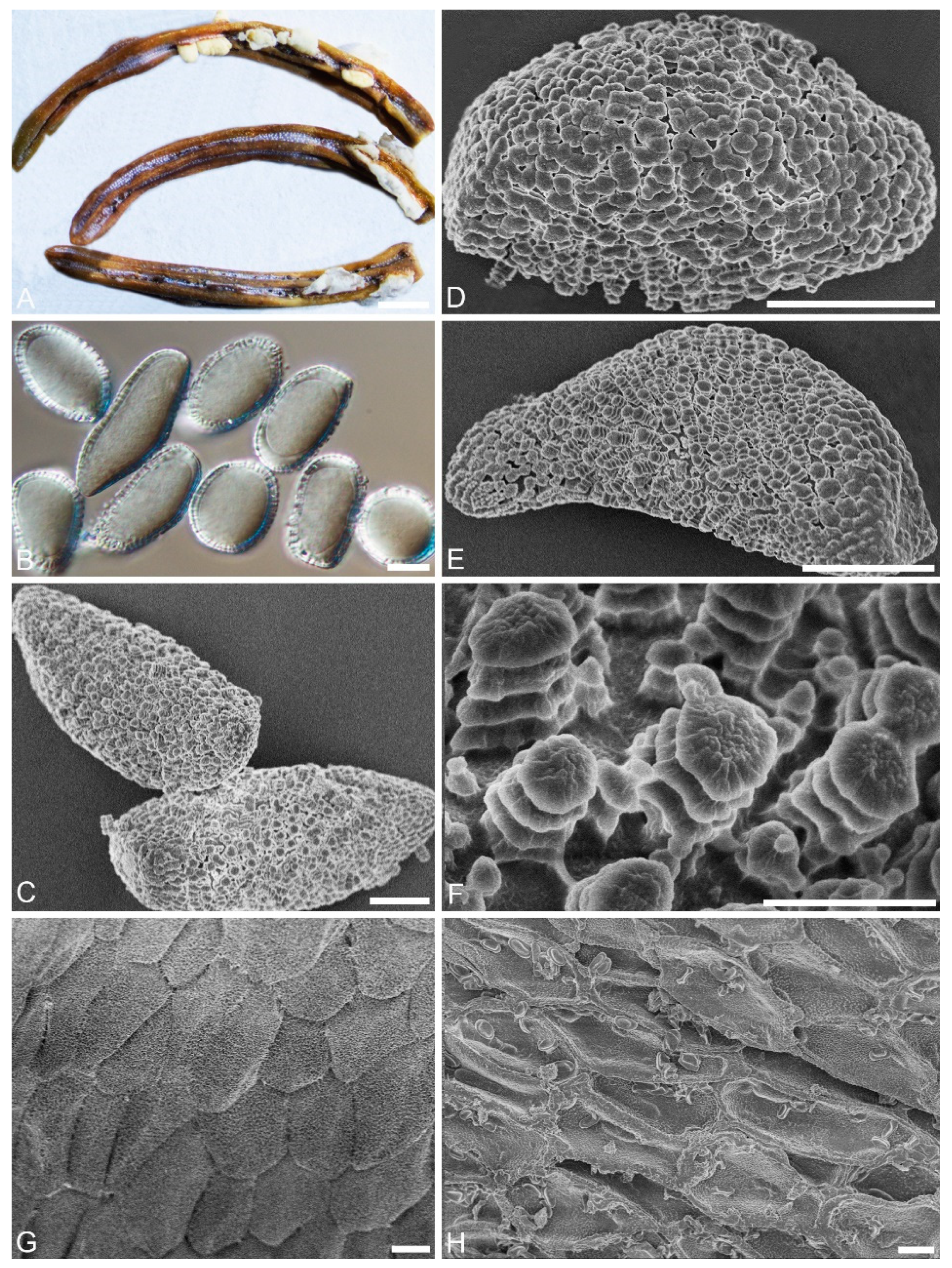
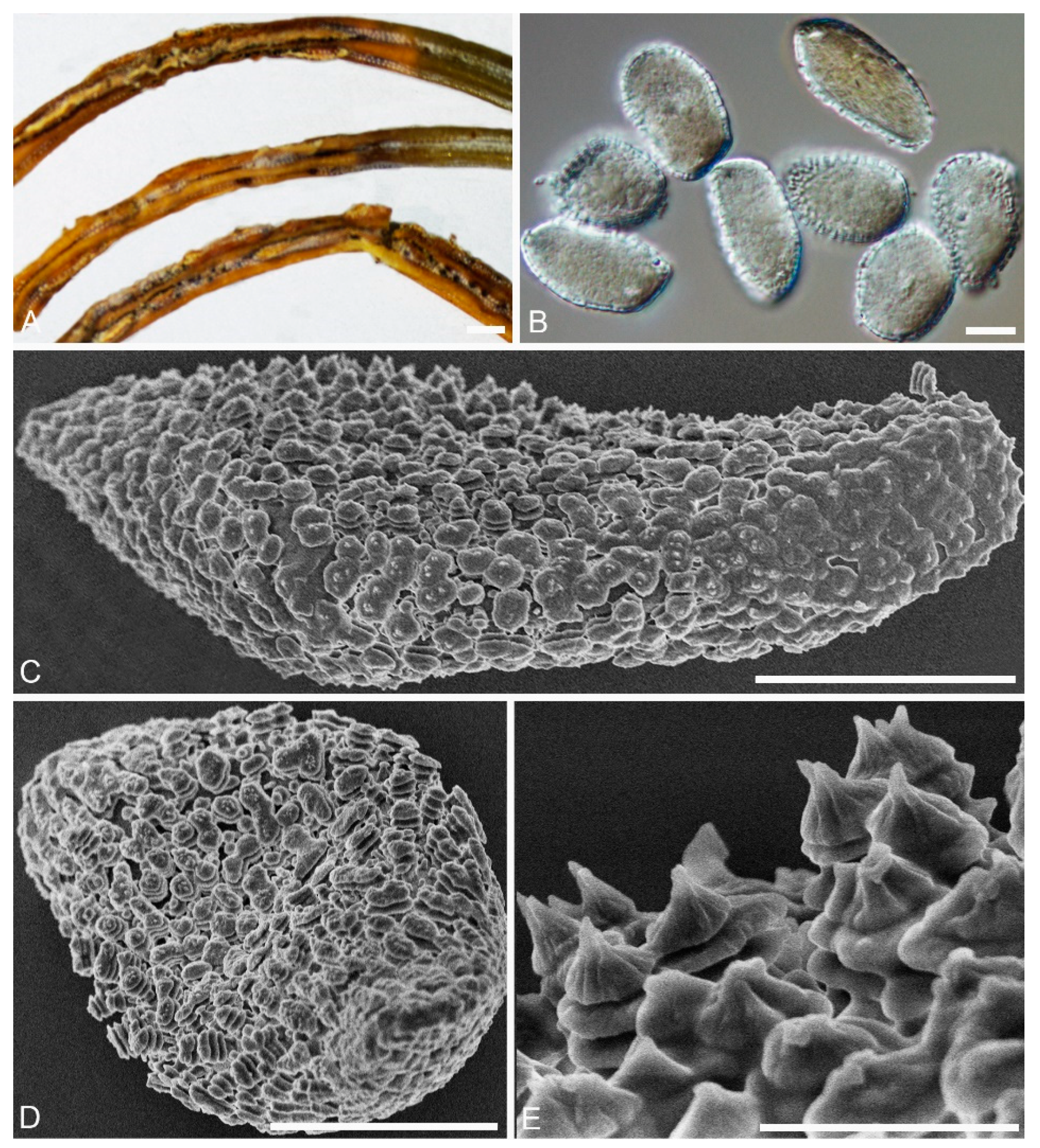
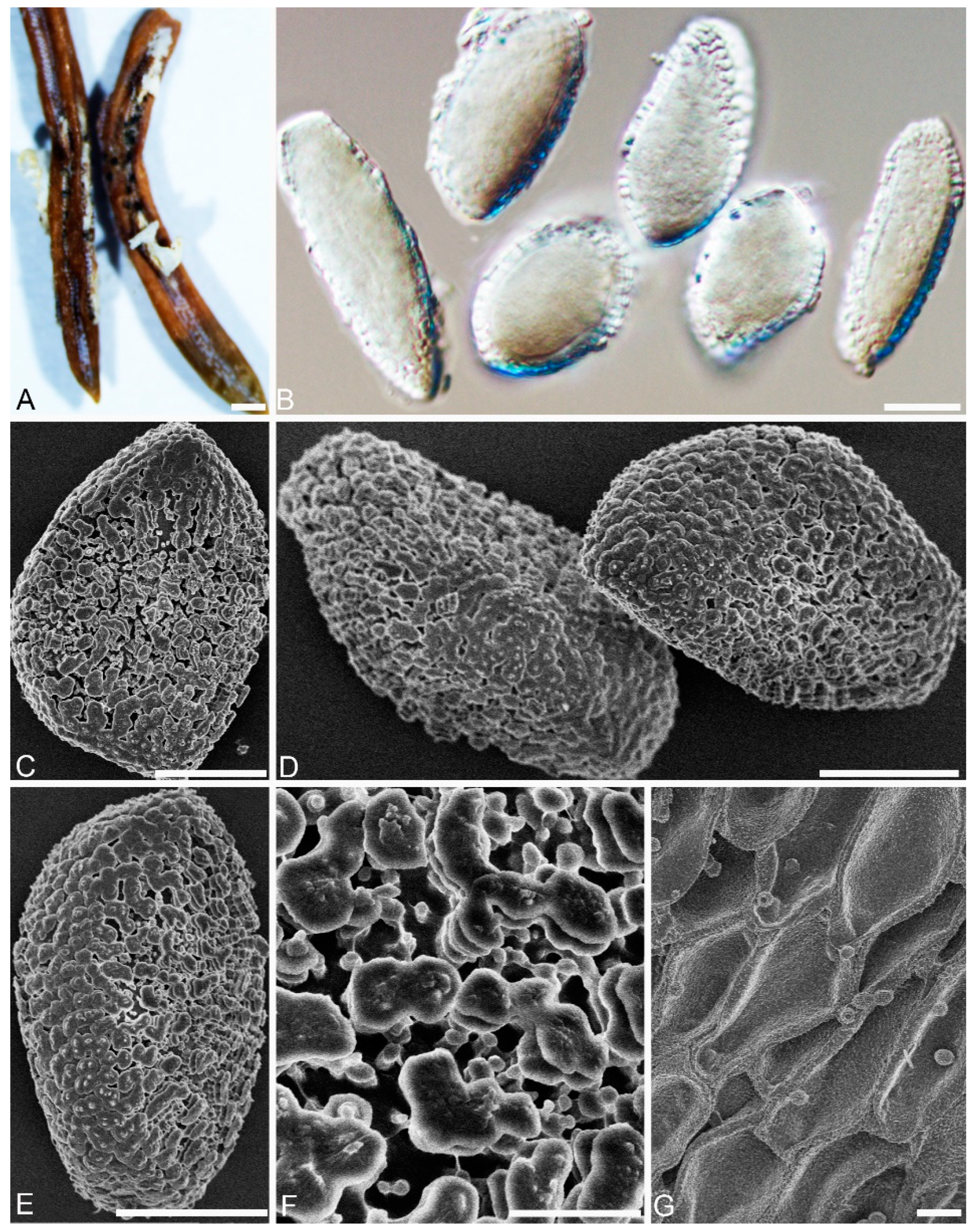
2.2. Morphological Analyses
2.3. DNA Extraction
2.4. Phylogenetic Analyses
2.5. Species Delimitation Analyses
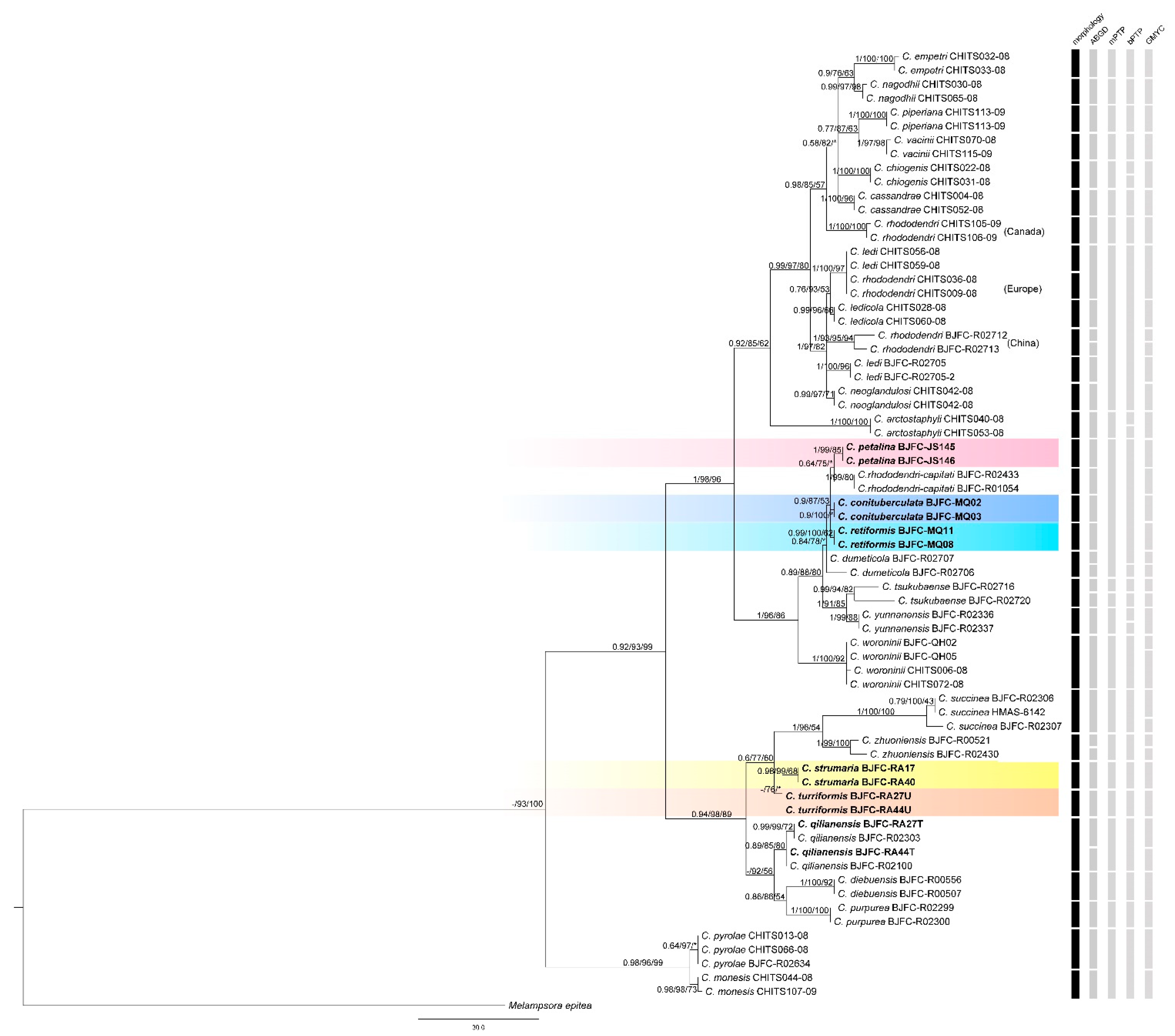
3. Results
3.1. Phylogenetic Relationships
3.2. Phylogenetic Relationships
3.3. Taxonomic Implications
| 1 Telium covered with transparent sheaths | 2 |
| 1 Telium not covered with transparent sheaths | 3 |
| 2 Aeciospores have unique echinulate warts | Chrysomyxa purpurea |
| 2 Urediniospore densely warted, one side covered by a shallowly warts, longitudinal cap with a ragged edge | Chrysomyxa forrestii |
| 3 Only the uredinial stage is found | 4 |
| 3 Uredinial and aecium stage are found | 6 |
| 4 Urediniospore have verrucose warts | Chrysomyxa tsukubaense |
| 4 Urediniospore have not verrucose warts | 5 |
| 5 Urediniospore with a narrow shallow cap at one or both ends, warts crowded, annulate, tower-like, with smooth and rounded tops | Chrysomyxa turriformis |
| 5 Urediniospore without cap | 6 |
| 6 Urediniospore not reticulate or smooth | 7 |
| 6 Urediniospore reticulate or smooth | 8 |
| 7 Urediniospore with densely crowded, narrow, spine-like warts | Chrysomyxa spinulospora |
| 7 Urediniospore with annulate, struma-like and fused warts | Chrysomyxa strumaria |
| 8 Urediniospore with broad, even, annualte warts | Chrysomyxa dumeticola |
| 8 Urediniospore with variable annulate warts interspersed with smaller warts | Chrysomyxa rhododendri-captitati |
| 9 Aeciospores not smooth or reticulate | 10 |
| 9 Aeciospores smooth or reticulate | 13 |
| 10 Aeciospore with nailheaded warts | Chrysomyxa dibuensis |
| 10 Aeciospore without nailheaded warts | 11 |
| 11Aeciospore with single echinae on peltate base; microcyclic | Chrysomyxa qilianensis |
| 11 Aeciospore without verrucose warts | 12 |
| 12 Aeciospore with two annuli warts, top of warts smooth and flat | Chrysomyxa pyrolae |
| 12 Aeciospore with three annuli warts, with a central spine on the top of warts | Chrysomyxa yunnanensis |
| 13 Aeciospore with cylindrical warts, with three annuli | 14 |
| 13 Aeciospore with cylindrical warts, without three annuli | 15 |
| 14 Aeciospore with a reticulated area at one or both ends, annulate warts generally consisting of three or four hemispherical layers, top of the layers in the shape of a conical cap, Aeciospore 24–31 × 15–20 μm | Chrysomyxa conituberculata |
| 14 Aeciospore with a reticulated area over the whole spore surface, warts crowded, annulate, with shallow bumps, smaller aeciospore 22–47 × 15–24 μm | Chrysomyxa retiformis |
| 15 Aeciospore with annulate warts, with 1–2 annuli, aeciospore 15–43×1439μm | Chrysomyxa woroninii |
| 15 Aeciospore with a reticulated area at one or both ends, warts cylindrical with thin basal connections, annulate, with petaloid tops, aeciospore 22–27 × 17–22 μm | Chrysomyxa petalina |
| 16 Aeciospore with a cap | 17 |
| 16 Aeciospore without a cap | 19 |
| 17 Aeciospore without a smooth cap | 18 |
| 17 Aeciospore with longitudinal smooth cap, with a broken, fissured edge | Chrysomyxa zhuoniensis |
| 18 Aeciospore with narrow cap, with longitudinal edge | 19 |
| 18 Aeciospore with broad longitudinal cap, with cylindrical warts with smooth or rough tops | Chrysomyxa succinea |
| Aeciospore with narrow warted groove, outer surface of aecial peridium concave | Chrysomyxa ledi |
| 19 Aeciospore with longitudinal smoother area with irregular bumps, inner surface of aecial peridium warted | Chrysomyxa rhododendri |
4. Discussion
4.1. Comparison of Species Delimitation Methods
4.2. Species Boundaries
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kirk, P.M.; Cannon, P.F.; Minter, D. Dictionary of the Fungi, 10th ed.; CABI: Wallingford, UK, 2008. [Google Scholar]
- Aime, M.C.; Mctaggart, A.R. A higher-rank classification for rust fungi, with notes on genera. Fungal Syst. Evol. 2020, 7, 21–47. [Google Scholar] [CrossRef]
- Zhimin, C.; Zhenqi, L. The New Records of Rus t Fungi from China. J. Northwest For. Univ. 1999, 14, 45–52. [Google Scholar]
- Cummins, G.; Hiratsuka, Y. Illustrated Genera of Rust Fungi, 3rd ed.; The American Phytopathological Society Press: St. Paul, MN, USA, 2003. [Google Scholar]
- Petersen, R.H. The rust fungi life cycle. Bot. Rev. 1974, 40, 453–513. [Google Scholar] [CrossRef]
- Ramsbottom, J. Some notes on the history of the classification of the Uredinales. Trans. Br. Mycol. Soc. 1912, 4, 77–105. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Sato, S. Morphology of aecia of the rust fungi. Trans. Br. Mycol. Soc. 1985, 85, 223–238. [Google Scholar] [CrossRef]
- Bennett, C.; Aime, M.C.; Newcombe, G. Molecular and pathogenic variation within Melampsora on Salix in western North America reveals numerous cryptic species. Mycologia 2011, 103, 1004–1018. [Google Scholar] [CrossRef] [PubMed]
- Berndt, R. Taxonomic revision of Endoraecium digitatum (rust fungi, Uredinales) with description of four new species from Australia and Hawaii. Mycol. Prog. 2011, 10, 497–517. [Google Scholar] [CrossRef]
- Beenken, L.; Zoller, S.; Berndt, R. Rust fungi on Annonaceae II: The genus Dasyspora Berk. & M.a. Curtis. Mycologia 2012, 104, 659–681. [Google Scholar]
- Cao, B.; Haelewaters, D.; Schoutteten, N.; Begerow, D.; Boekhout, T.; Giachini, A.J.; Gorjón, S.P.; Gunde-Cimerman, N.; Hyde, K.D.; Kemler, M.; et al. Delimiting species in Basidiomycota: A review. Fungal Divers. 2021, 109, 181–237. [Google Scholar] [CrossRef]
- Crane, P.E. Morphology, taxonomy, and nomenclature of the Chrysomyxa ledi complex and related rust fungi on spruce and Ericaceae in North America and Europe. Can. J. Bot. 2001, 79, 957–982. [Google Scholar] [CrossRef]
- Cao, J.; Tian, C.; Liang, Y.; You, C. A new rust species of Diaphanopellis on Rhododendron oreodoxa from Southern China. Phytotaxa 2017, 309, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Tian, C.; Liang, Y.; You, C. Two new Chrysomyxa rust species on the endemic plant, Picea asperata in western China, and expanded description of C. Succinea. Phytotaxa 2017, 292, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Feau, N.; Vialle, A.; Allaire, M.; Maier, W.; Hamelin, R.C. DNA barcoding in the rust genus Chrysomyxa and its implications for the phylogeny of the genus. Mycologia 2011, 103, 1250–1266. [Google Scholar] [CrossRef] [PubMed]
- You, C.J.; Yang, L.J.; Tian, C.M. Resolving the phylogenetic position of Caeoma spp. That infect Rhododendron and Chrysomyxa from China. Mycol. Prog. 2019, 18, 1285–1299. [Google Scholar] [CrossRef]
- Maier, W.; Begerow, D.; Weiß, M.; Oberwinkler, F. Phylogeny of the rust fungi: An approach using nuclear large subunit ribosomal DNA sequences. Can. J. Bot. 2003, 81, 12–23. [Google Scholar] [CrossRef]
- Berndt, R. Chrysomyxa rust: Morphology and ultrastructure of Dhaustoria, uredinia and telia. Can. J. Bot. 1999, 77, 1469–1484. [Google Scholar] [CrossRef]
- Crane, P.E. Rust fungi on rhododendrons in Asia: Diaphanopellis forrestii gen. Et sp. Nov., New species of Caeoma, and expanded descriptions of Chrysomyxa dietelii and C. Succinea. Mycologia 2005, 97, 534–548. [Google Scholar] [CrossRef]
- Sánchez-Restrepo, A.F.; Chifflet, L.; Confalonieri, V.A.; Tsutsui, N.D.; Pesquero, M.A.; Calcaterra, L.A. A Species delimitation approach to uncover cryptic species in the South American fire ant decapitating flies (Diptera: Phoridae: Pseudacteon). PLoS ONE 2020, 15, e236086. [Google Scholar]
- Sato, H.; Ohta, R.; Murakami, N. Molecular prospecting for cryptic species of the Hypholoma fasciculare complex: Toward the effective and practical delimitation of cryptic macrofungal species. Sci. Rep.-UK 2020, 10, 13224. [Google Scholar] [CrossRef] [PubMed]
- Maharachchikumbura, S.S.N.; Chen, Y.; Ariyawansa, H.A.; Hyde, K.D.; Haelewaters, D.; Perera, R.H.; Samarakoon, M.C.; Wanasinghe, D.N.; Bustamante, D.E.; Liu, J.; et al. Integrative approaches for species delimitation in Ascomycota. Fungal Divers. 2021, 109, 155–179. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-rate Poisson Tree Processes for single-locus species delimitation under Maximum Likelihood and Markov Chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Blair, C.; Bryson, R.W. Cryptic diversity and discordance in single-locus species delimitation methods within horned lizards (Phrynosomatidae: Phrynosoma). Mol. Ecol. Resour. 2017, 17, 1168–1182. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, E.P.; Nicholson, K.E.; Luque-Montes, I.R.; Kohler, G.; Cerrato-Mendoza, C.A.; Medina-Flores, M.; Wilson, L.D.; Townsend, J.H. Cryptic diversity, but to what extent? Discordance between Single-Locus species delimitation methods within mainland anoles (Squamata: Dactyloidae) of northern central America. Front Genet. 2019, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Savile, D. Chrysomyxa in North America—additions and corrections. Can. J. Bot. 1955, 33, 487–496. [Google Scholar] [CrossRef]
- Ziller, W. The Tree Rusts of Western Canada; Canadian Forestry Service: Ottawa, ON, Canada, 1975. [Google Scholar]
- Zhuang, J.; Zheng, X. Known species of the genus Chrysomyxa Unger (Uredinales, Chrysomyxaceae) in China. J. Xichang Univ. (Nat. Sci. Ed.) 2017, 31, 1–9, 26. [Google Scholar]
- Yang, T. Phylogenetic and Taxonomic Studies of Pucciniastrum s.l. Ph.D. Thesis, Beijing Forestry University, Beijing, China, 2015. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods) Version 4.0b10; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Nylander, J.A.A.; Wilgenbusch, J.C.; Warren, D.L.; Swofford, D.L. AWTY (are we there yet?): A system for graphical exploration of MCMC convergence in Bayesian phylogenetics. Bioinformatics 2008, 24, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Gascuel, O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating Maximum-Likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Villota, E.Y.; Carmignotto, A.P.; Brandão, M.V.; Percequillo, A.R.; Silva, M.J.D.J. Systematics of the genus Oecomys (Sigmodontinae: Oryzomyini): Molecular phylogenetic, cytogenetic and morphological approaches reveal cryptic species. Zool. J. Linn. Soc.-Lond. 2017, 184, 182–210. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Hnert, D.K.; Vaughan, T.; Wu, C.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar]
- Zhao, P.; Liu, F.; Li, Y.; Cai, L. Inferring phylogeny and speciation of Gymnosporangium species and their coevolution with host plants. Sci. Rep.-UK 2016, 6, 29339. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 5 September 2021).
- Ahrens, D.; Fujisawa, T.; Krammer, H.; Eberle, J.; Fabrizi, S.; Vogler, A.P. Rarity and incomplete sampling in DNA-Based species delimitation. Syst. Biol. 2016, 65, 478–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, A.; Ling, C.; Ho, S.Y.W.; Zhu, C. Comparison of methods for molecular species delimitation across a range of speciation scenarios. Syst. Biol. 2018, 67, 830–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haelewaters, D.; De Kesel, A.; Pfister, D.H. Integrative taxonomy reveals hidden species within a common fungal parasite of ladybirds. Sci. Rep.-UK 2018, 8, 15966. [Google Scholar] [CrossRef] [PubMed]
- Haelewaters, D.; Pfister, D.H. Morphological species of Gloeandromyces (Ascomycota, laboulbeniales) evaluated using single-locus species delimitation methods. Fungal Syst. Evol. 2019, 3, 19–34. [Google Scholar] [CrossRef] [Green Version]
- De Lange, R.; Adamčík, S.; Adamčíkova, K.; Asselman, P.; Borovička, J.; Delgat, L.; Hampe, F.; Verbeken, A. Enlightening the black and white: Species delimitation and UNITE species hypothesis testing in the Russula albonigra species complex. IMA Fungus 2021, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Sklenář, F.; Jurjević, Ž.; Houbraken, J.; Kolařík, M.; Arendrup, M.C.; Jørgensen, K.M.; Siqueira, J.P.Z.; Gené, J.; Yaguchi, T.; Ezekiel, C.N.; et al. Re-examination of species limits in Aspergillus section Flavipedes using advanced species delimitation methods and description of four new species. Stud. Mycol. 2021, 99, 100120. [Google Scholar] [CrossRef]
- Guarnizo, C.E.; Paz, A.; Muñoz-Ortiz, A.; Flechas, S.V.; Méndez-Narváez, J.; Crawford, A.J. DNA barcoding survey of anurans across the eastern cordillera of colombia and the impact of the andes on cryptic diversity. PLoS ONE 2015, 10, e127312. [Google Scholar]
- Dellicour, S.; Flot, J. The hitchhiker’s guide to single-locus species delimitation. Mol. Ecol. Resour. 2018, 18, 1234–1246. [Google Scholar] [CrossRef]
- Boissin, E.; Hoareau, T.B.; Paulay, G.; Bruggemann, J.H. DNA barcoding of reef brittle stars (Ophiuroidea, Echinodermata) from the southwestern Indian Ocean evolutionary hot spot of biodiversity. Ecol. Evol. 2017, 7, 11197–11203. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; Mcguire, J.A.; Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 2012, 27, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Miralles, A.; Vences, M.; Fontaneto, D. New metrics for comparison of taxonomies reveal striking discrepancies among species delimitation methods in Madascincus lizards. PLoS ONE 2013, 8, e68242. [Google Scholar]
- Talavera, G.; Lukhtanov, V.A.; Rieppel, L.; Pierce, N.E.; Vila, R. In the shadow of phylogenetic uncertainty: The recent diversification of Lysandra butterflies through chromosomal change. Mol. Phylogenet. Evol. 2013, 69, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, R.; Montero-Mendieta, S.; Simó-Riudalbas, M.; Sindaco, R.; Santos, X.; Fasola, M.; Llorente, G.; Razzetti, E.; Carranza, S. Unexpectedly high levels of cryptic diversity uncovered by a complete DNA barcoding of reptiles of the socotra archipelago. PLoS ONE 2016, 11, e149985. [Google Scholar]
- Kornilios, P.; Kumlutaş, Y.; Lymberakis, P.; Ilgaz, Ç. Cryptic diversity and molecular systematics of the Aegean Ophiomorus skinks (Reptilia: Squamata), with the description of a new species. J. Zool. Syst. Evol. Res. 2018, 56, 364–381. [Google Scholar] [CrossRef]
- Esselstyn, J.A.; Evans, B.J.; Sedlock, J.L.; Khan, F.A.A.; Heaney, L.R. Single-locus species delimitation: A test of the mixed Yule–coalescent model, with an empirical application to Philippine round-leaf bats. Proc. R. Soc. B 2012, 279, 3678–3686. [Google Scholar] [CrossRef]
- Tang, C.Q.; Humphreys, A.M.; Fontaneto, D.; Barraclough, T.G. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 2014, 5, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y. A new spruce needle rust fungus. Acta Mycol. Sin. 1987, 6, 86–88. [Google Scholar]
- García-Guzmán, G.; Wennström, A. Interactions between two rust fungi and their host plant Anemone nemorosa. Ecography 2001, 24, 25–32. [Google Scholar] [CrossRef]
- Jörstad, I. Uromyces on Trifolium repens. Nytt. Mag. Bot. 1967, 14, 19–30. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NumOTU a | Iss b | Iss.cSym c | Psym d | Iss.cAsyme | Pasym f |
|---|---|---|---|---|---|
| 4 | 0.430 | 0.826 | 0.000 | 0.794 | 0.000 |
| 8 | 0.425 | 0.797 | 0.000 | 0.693 | 0.000 |
| 16 | 0.491 | 0.780 | 0.000 | 0.588 | 0.004 |
| 32 | 0.558 | 0.759 | 0.000 | 0.463 | 0.020 |
| ABGD a | mPTP b | bPTP c | GMYC d | |
|---|---|---|---|---|
| Nmatche | 28 | 20 | 20 | 27 |
| Ndelimitedf | 33 | 21 | 25 | 35 |
| Match ratio | 0.90 | 0.80 | 0.74 | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Tsui, C.K.M.; You, C. Cryptic Species Diversity and Phylogenetic Relationship in the Rust Genus Chrysomyxa from China. J. Fungi 2022, 8, 83. https://doi.org/10.3390/jof8010083
Wang R, Tsui CKM, You C. Cryptic Species Diversity and Phylogenetic Relationship in the Rust Genus Chrysomyxa from China. Journal of Fungi. 2022; 8(1):83. https://doi.org/10.3390/jof8010083
Chicago/Turabian StyleWang, Rui, Clement K. M. Tsui, and Chongjuan You. 2022. "Cryptic Species Diversity and Phylogenetic Relationship in the Rust Genus Chrysomyxa from China" Journal of Fungi 8, no. 1: 83. https://doi.org/10.3390/jof8010083