
Global Transcriptome Profile of the Oleaginous Yeast Saitozyma podzolica DSM 27192 Cultivated in Glucose and Xylose


Abstract
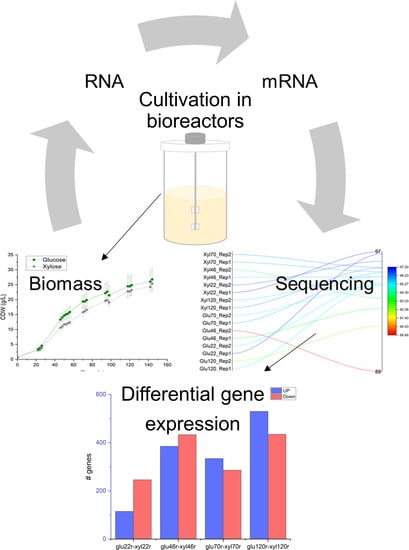
:
1. Introduction
2. Materials and Methods
2.1. Yeast Strain and Experimental Set Up
2.2. RNA Isolation, Library Construction and Sequencing
2.3. RNA-Seq Data Analyses
2.4. Functional Analysis of Expressed Genes
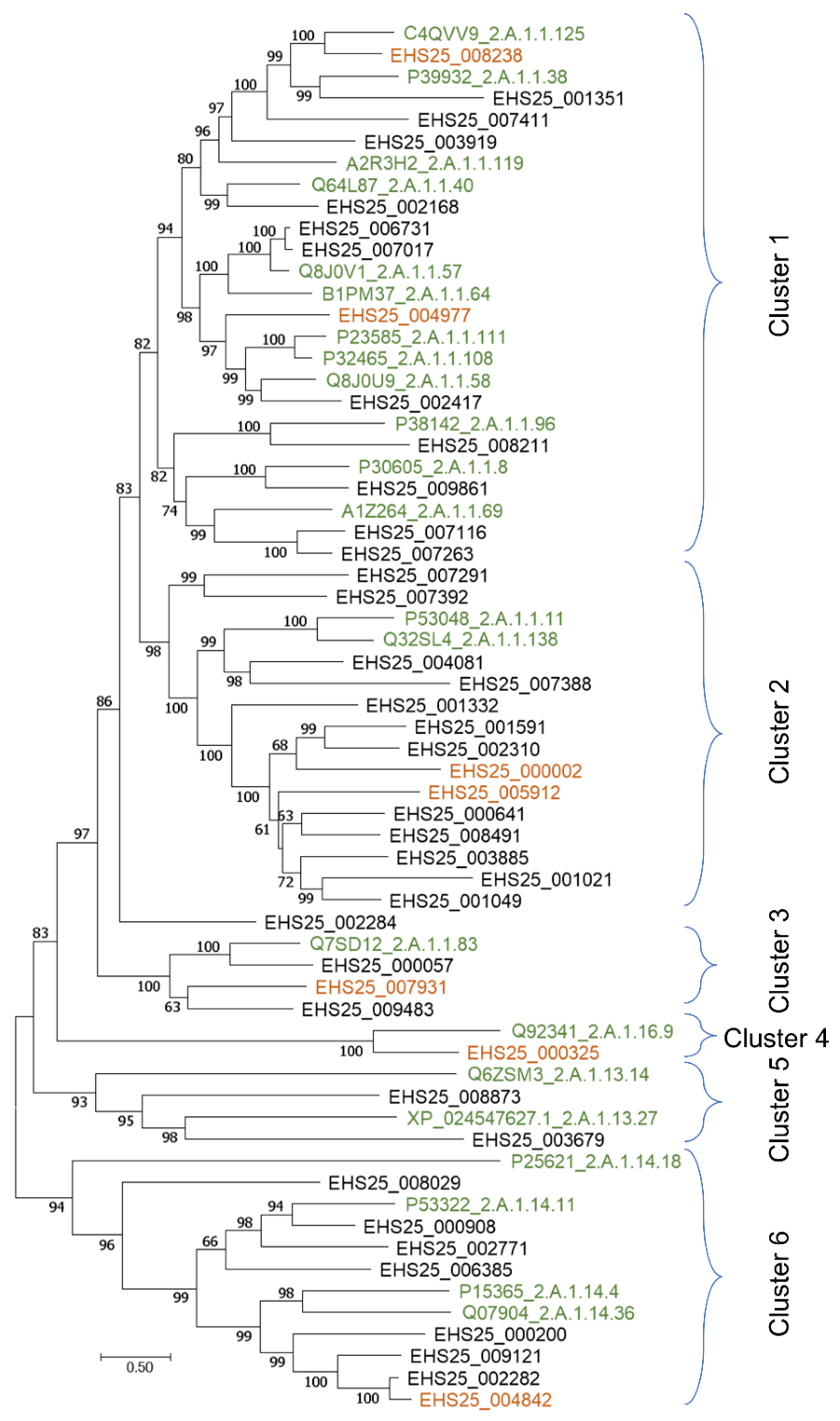
2.5. Phylogenetic Analysis of Sugar Transport Proteins
3. Results and Discussion
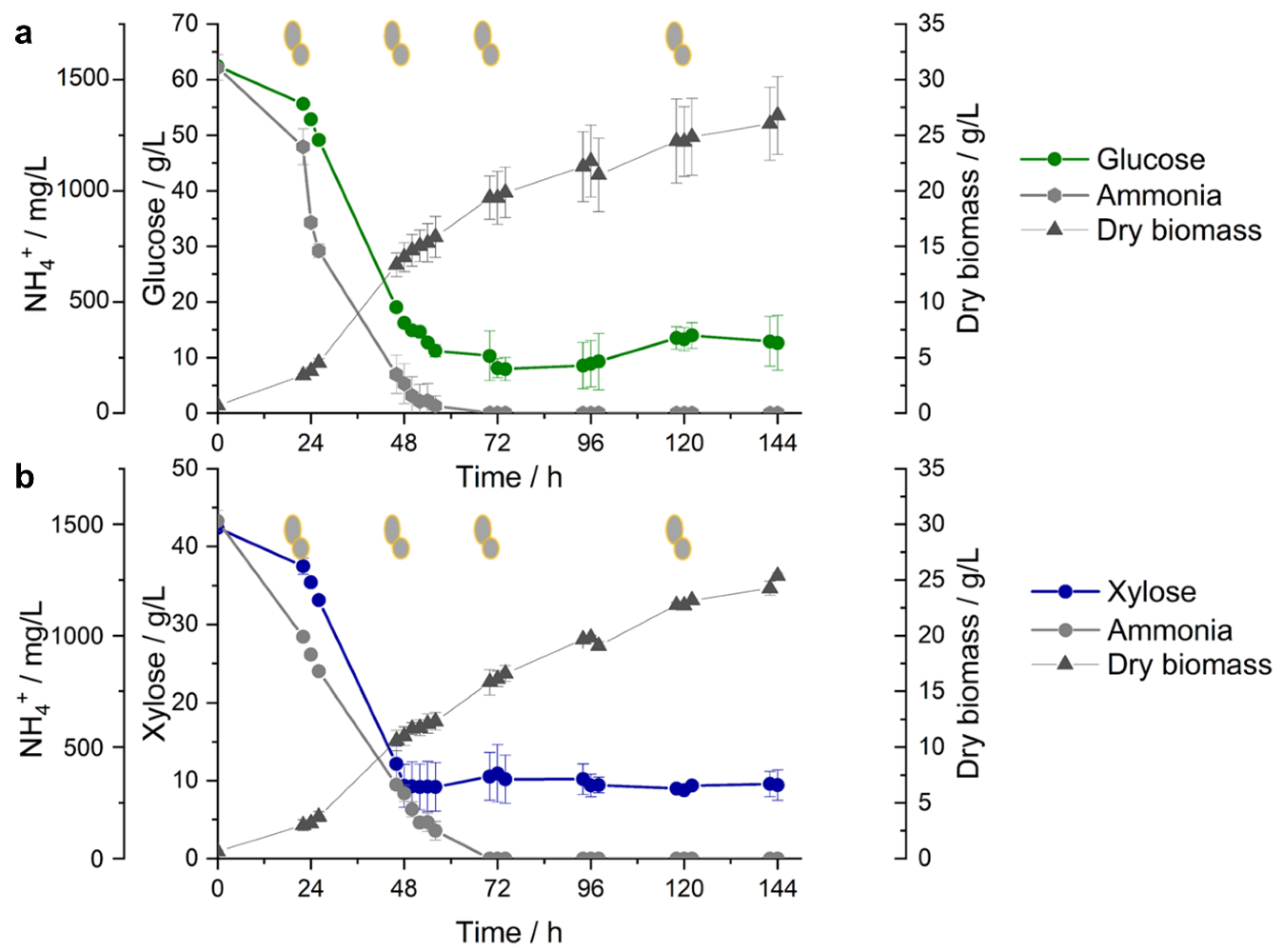
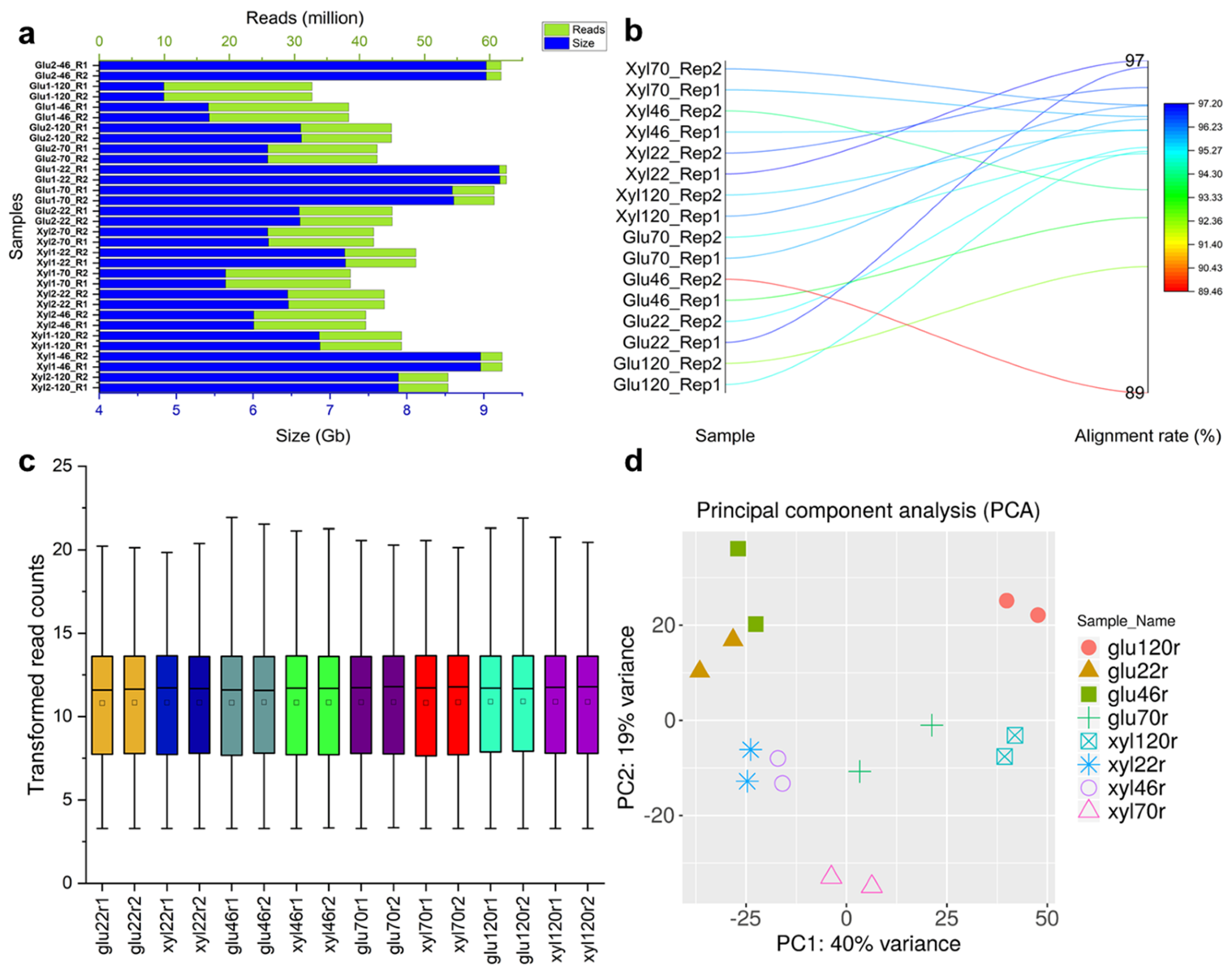
3.1. Overview of Yeast Cultivation and RNA-Seq
3.2. Differential Gene Expression during Initial Growth under Glucose and Xylose in DSM 27192
3.3. Distinct Profiles of Putative Sugar Transporter Genes in Saitozyma Podzolica DSM 27192 Grown on Glucose and Xylose
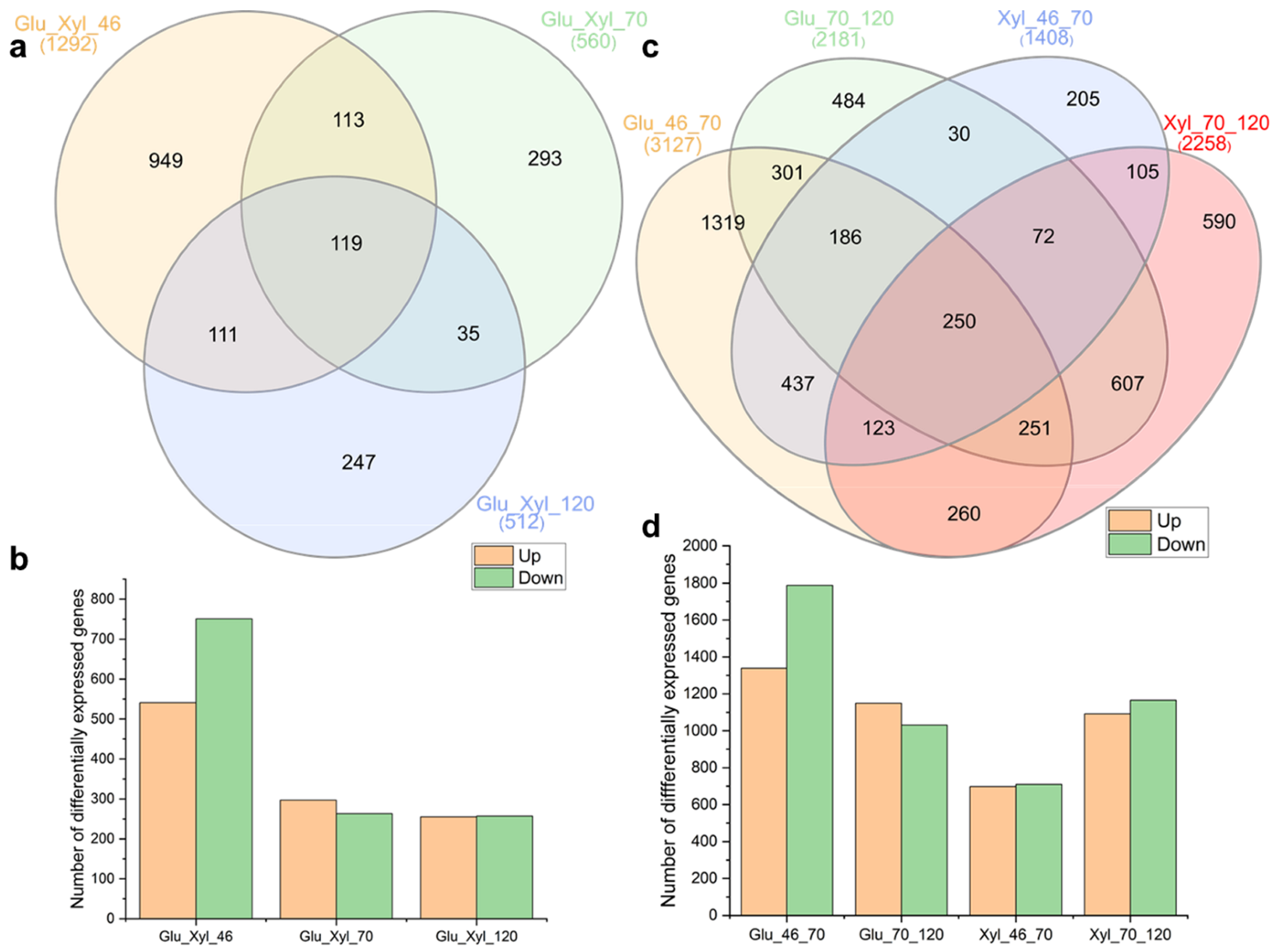
3.4. Comparison of Transcriptome Profiles of Saitozyma Podzolica DSM 27192 during Continuous Feeding on Glucose and Xylose
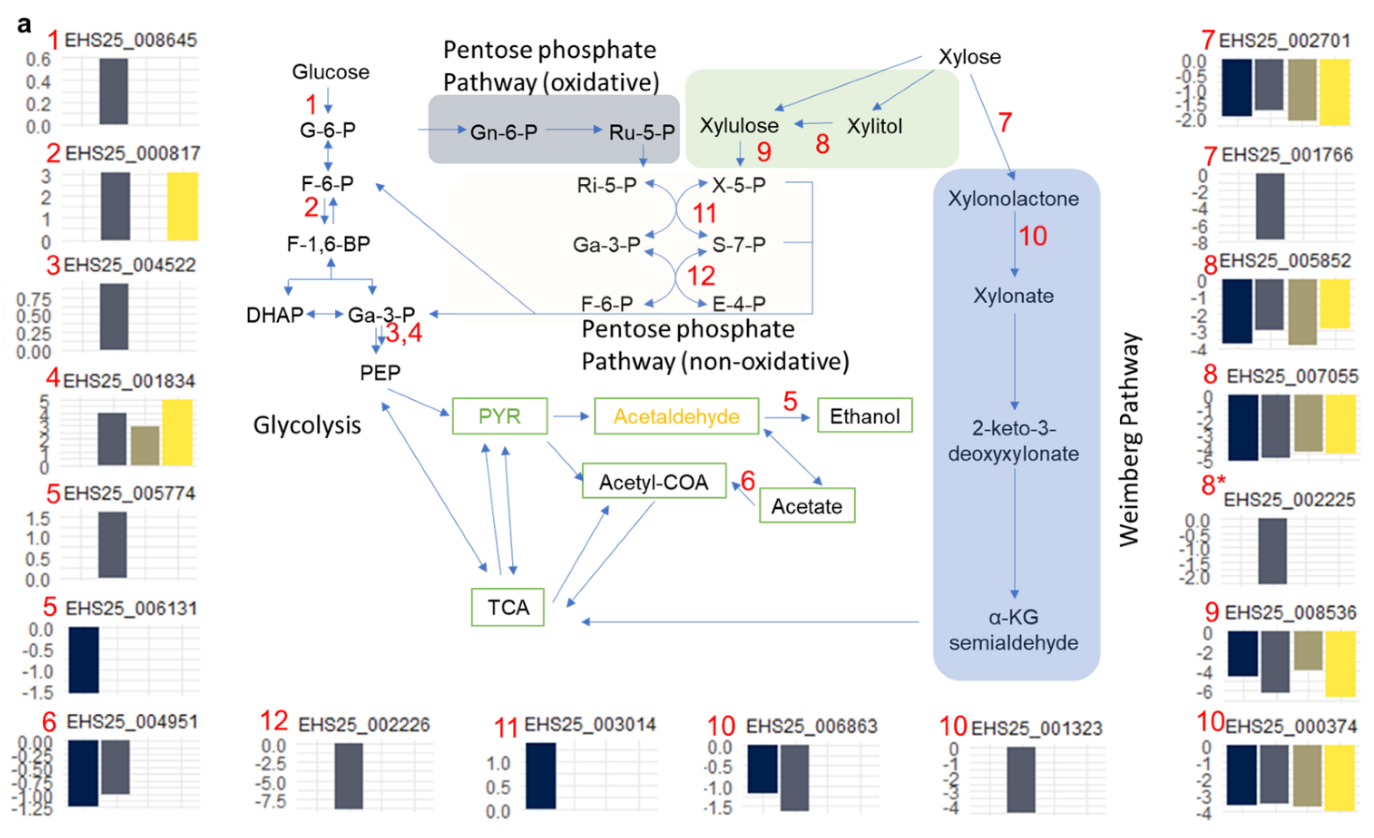
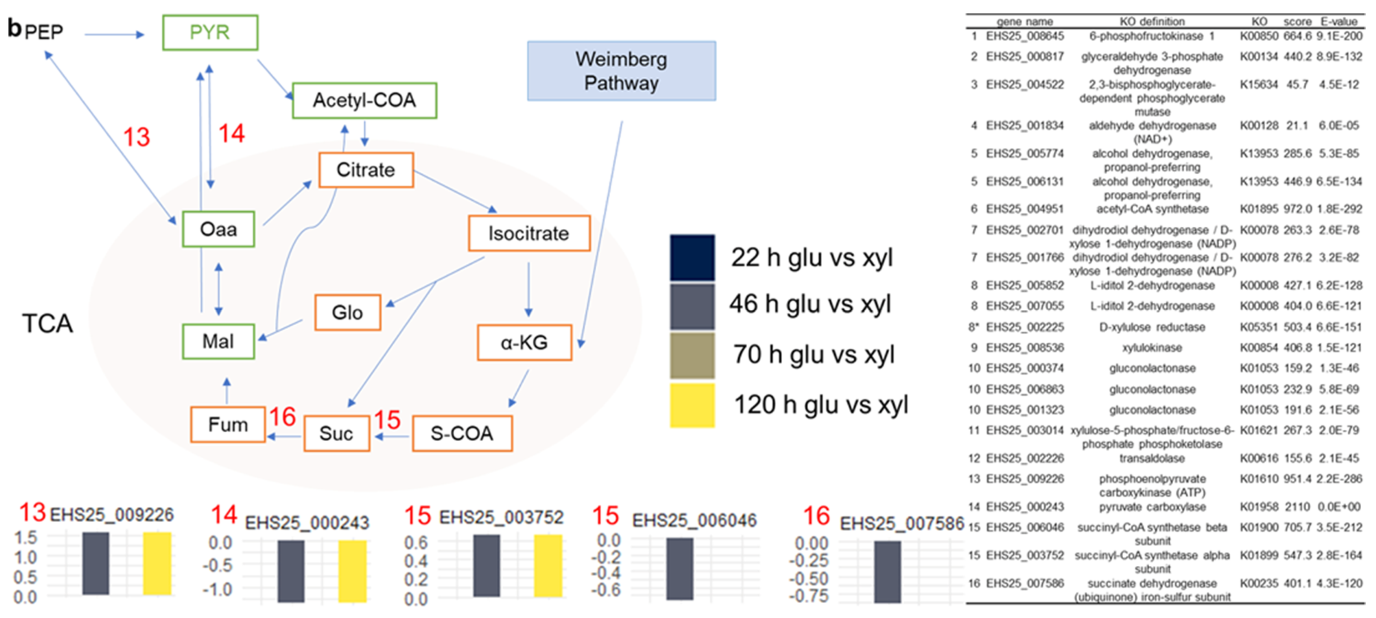
3.5. Temporal Dynamics of S. podzolica DSM 27192 Genes Grown under Glucose and Xylose
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Prospects 2019: Highlights; United Nations: New York, NY, USA, 2019; p. 40. [Google Scholar]
- Ratledge, C. Fatty acid biosynthesis in microorganisms being used for single cell oil production. Biochimie 2004, 86, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, A.; Sannino, F.; Addorisio, V.; Pirozzi, D. Microbial conversion of olive oil mill wastewaters into lipids suitable for biodiesel production. J. Agric. Food Chem. 2010, 58, 8630–8635. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Nip, S.; Shim, H. Enhancement of lipid productivity of Rhodosporidium toruloides in distillery wastewater by increasing cell density. Bioresour. Technol. 2013, 146, 301–309. [Google Scholar] [CrossRef]
- Kot, A.; Błażejak, S.; Kurcz, A.; Bryś, J.; Gientka, I.; Bzducha-Wróbel, A.; Maliszewska, M.; Reczek, L. Effect of initial pH of medium with potato wastewater and glycerol on protein, lipid and carotenoid biosynthesis by Rhodotorula glutinis. Electron. J. Biotechnol. 2017, 27, 25–31. [Google Scholar] [CrossRef]
- Li, Q.; Du, W.; Liu, D. Perspectives of microbial oils for biodiesel production. Appl. Microbiol. Biotechnol. 2008, 80, 749–756. [Google Scholar] [CrossRef]
- Ageitos, J.M.; Vallejo, J.; Veiga-Crespo, P.; Villa, T.G. Oily yeasts as oleaginous cell factories. Appl. Microbiol. Biotechnol. 2011, 90, 1219–1227. [Google Scholar] [CrossRef]
- Aliyu, H.; Gorte, O.; Zhou, X.; Neumann, A.; Ochsenreither, K. In silico proteomic analysis provides insights into phylogenomics and plant biomass deconstruction potentials of the tremelalles. Front. Bioeng. Biotechnol. 2020, 8, 226. [Google Scholar] [CrossRef]
- Aliyu, H.; Gorte, O.; de Maayer, P.; Neumann, A.; Ochsenreither, K. Genomic insights into the lifestyles, functional capacities and oleagenicity of members of the fungal family Trichosporonaceae. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gorte, O.; Kugel, M.; Ochsenreither, K. Optimization of carbon source efficiency for lipid production with the oleaginous yeast Saitozyma podzolica DSM 27192 applying automated continuous feeding. Biotechnol. Biofuels 2020, 13, 1–17. [Google Scholar] [CrossRef]
- Meesters, P.A.E.P.; Huijberts, G.N.M.; Eggink, G. High-cell-density cultivation of the lipid accumulating yeast Cryptococcus curvatus using glycerol as a carbon source. Appl. Microbiol. Biotechnol. 1996, 45, 575–579. [Google Scholar] [CrossRef]
- Fei, Q.; O’Brien, M.; Nelson, R.; Chen, X.; Lowell, A.; Dowe, N. Enhanced lipid production by Rhodosporidium toruloides using different fed-batch feeding strategies with lignocellulosic hydrolysate as the sole carbon source. Biotechnol. Biofuels 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poontawee, R.; Limtong, S. Feeding strategies of two-stage fed-batch cultivation processes for microbial lipid production from sugarcane top hydrolysate and crude glycerol by the oleaginous red yeast Rhodosporidiobolus fluvialis. Microorganisms 2020, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gírio, F.; Fonseca, C.; Carvalheiro, F.; Duarte, L.; Marques, S.; Lukasik, R. Hemicelluloses for fuel ethanol: A review. Bioresour. Technol. 2010, 101, 4775–4800. [Google Scholar] [CrossRef] [PubMed]
- Schulze, I.; Hansen, S.; Großhans, S.; Rudszuck, T.; Ochsenreither, K.; Syldatk, C.; Neumann, A. Characterization of newly isolated oleaginous yeasts—Cryptococcus podzolicus, Trichosporon porosum and Pichia segobiensis. AMB Express 2014, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Gorte, O.; Chen, L.; Zhang, W.; Dong, W.; Ma, J.; Jiang, M.; Xin, F.; Ochsenreither, K. Co-production of single cell oil and gluconic acid using oleaginous Cryptococcus podzolicus DSM 27192. Biotechnol. Biofuels 2019, 12, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorte, O.; Hollenbach, R.; Papachristou, I.; Steinweg, C.; Silve, A.; Frey, W.; Syldatk, C.; Ochsenreither, K. Evaluation of downstream processing, extraction, and quantification strategies for single cell oil produced by the oleaginous yeasts Saitozyma podzolica DSM 27192 and Apiotrichum porosum DSM 27194. Front. Bioeng. Biotechnol. 2020, 8, 355. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Aliyu, H.; Gorte, O.; Neumann, A.; Ochsenreither, K. Draft genome sequence of the oleaginous yeast Saitozyma podzolica (syn. Cryptococcus podzolicus) DSM 27192. Microbiol. Resour. Announc. 2019, 8, e01676-18. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Son, E.W.; Yao, R. iDEP: An integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinform. 2018, 19, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Nueda, M.J.; Tarazona, S.; Conesa, A. Next maSigPro: Updating maSigPro bioconductor package for RNA-seq time series. Bioinformatics 2014, 30, 2598–2602. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Nueda, M.J.; Ferrer, A.; Talón, M. maSigPro: A method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics 2006, 22, 1096–1102. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Derbyshire, M.K.; Gonzales, N.R.; Lu, S.; Chitsaz, F.; Geer, L.Y.; Geer, R.C.; He, J.; Gwadz, M.; Hurwitz, D.I.; et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015, 43, D222–D226. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Saier, M.H.; Reddy, V.S.; Moreno-Hagelsieb, G.; Hendargo, K.J.; Zhang, Y.; Iddamsetty, V.; Lam, K.J.K.; Tian, N.; Russum, S.; Wang, J.; et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2021, 49, D461–D467. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.-M.; Taly, J.-F.; Notredame, C. T-coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Iwanicki, N.S.; Júnior, I.D.; Eilenberg, J.; De Fine Licht, H.H. Comparative RNAseq analysis of the insect-pathogenic fungus Metarhizium anisopliae reveals specific transcriptome signatures of filamentous and yeast-like development. G3 Genes Genomes Genet. 2020, 10, 2141–2157. [Google Scholar] [CrossRef]
- Hess, J.; Balasundaram, S.V.; Bakkemo, R.I.; Drula, E.; Henrissat, B.; Högberg, N.; Eastwood, D.; Skrede, I. Niche differentiation and evolution of the wood decay machinery in the invasive fungus Serpula lacrymans. ISME J. 2021, 15, 592–604. [Google Scholar] [CrossRef]
- Sturm, L.; Geißel, B.; Martin, R.; Wagener, J. Differentially regulated transcription factors and ABC transporters in a mitochondrial dynamics mutant can alter azole susceptibility of Aspergillus fumigatus. Front. Microbiol. 2020, 11, 1017. [Google Scholar] [CrossRef]
- Zhan, C.; Bai, Z.; Wang, S.; Sun, Y.; Dai, X.; Liu, X.; Harvey, L.; McNeil, B.; Yang, Y. The Pichia pastoris transmembrane protein GT1 is a glycerol transporter and relieves the repression of glycerol on AOX1 expression. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, Y.; Zhan, C.; Zhang, Z.; Liu, X.; Liu, H.; Bai, Z. Transcriptional analysis of impacts of glycerol transporter 1 on methanol and glycerol metabolism in Pichia pastoris. FEMS Yeast Res. 2017, 18, 18. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.; van Voorst, F.; Martins, A.; Neves, L.; Oliveira, R.; Kielland-Brandt, M.; Lucas, C.; Brandt, A. A member of the sugar transporter family, Stl1p is the glycerol/H+ symporter in Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 2068–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobre, A.; Lucas, C.; Leão, C. Transport and utilization of hexoses and pentoses in the halotolerant yeast Debaryomyces hansenii. Appl. Environ. Microbiol. 1999, 65, 3594–3598. [Google Scholar] [CrossRef] [Green Version]
- Jørgensen, T.R.; Vankuyk, P.A.; Poulsen, B.R.; Ruijter, G.J.G.; Visser, J.; Iversen, J.J.L. Glucose uptake and growth of glucose-limited chemostat cultures of Aspergillus niger and a disruptant lacking MstA, a high-affinity glucose transporter. Microbiology 2007, 153, 1963–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stasyk, O.G.; Maidan, M.M.; Stasyk, O.V.; van Dijck, P.; Thevelein, J.M.; Sibirny, A.A. Identification of Hexose transporter-like sensor HXS1 and functional hexose transporter HXT1 in the methylotrophic yeast Hansenula polymorpha. Eukaryot. Cell 2008, 7, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Viigand, K.; Tammus, K.; Alamäe, T. Clustering of MAL genes in Hansenula polymorpha: Cloning of the maltose permease gene and expression from the divergent intergenic region between the maltose permease and maltase genes. FEMS Yeast Res. 2005, 5, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, F.; Vidgren, V.; Ruohonen, L.; Gibson, B. Maltose and maltotriose utilisation by group I strains of the hybrid lager yeast Saccharomyces pastorianus. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Li, Y.; HamediRad, M.; Zhao, H. Directed evolution of a cellodextrin transporter for improved biofuel production under anaerobic conditions in Saccharomyces cerevisiae. Biotechnol. Bioeng. 2014, 111, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Vela-Corcía, D.; Srivastava, D.A.; Dafa-Berger, A.; Rotem, N.; Barda, O.; Levy, M. MFS transporter from Botrytis cinerea provides tolerance to glucosinolate-breakdown products and is required for pathogenicity. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zheng, Y.; Xiong, X.; Chen, S. Co-utilization of glucose, xylose and cellobiose by the oleaginous yeast Cryptococcus curvatus. Biomass Bioenergy 2014, 71, 340–349. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Zhang, X.; Hu, F.; Ryu, D.D.Y.; Bao, J. Screening of oleaginous yeast strains tolerant to lignocellulose degradation compounds. Appl. Biochem. Biotechnol. 2009, 159, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Belinchón, M.M.; Gancedo, J.M. Xylose and some non-sugar carbon sources cause catabolite repression in Saccharomyces cerevisiae. Arch. Microbiol. 2003, 180, 293–297. [Google Scholar] [CrossRef]
- Portnoy, T.; Margeot, A.; Seidl-Seiboth, V.; Le Crom, S.; Ben Chaabane, F.; Linke, R.; Seiboth, B.; Kubicek, C.P. Differential regulation of the cellulase transcription factors XYR1, ACE2, and ACE1 Trichoderma reesei strains producing high and low levels of cellulase. Eukaryot. Cell 2011, 10, 262–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alazi, E.; Ram, A.F.J. Modulating transcriptional regulation of plant biomass degrading enzyme networks for rational design of industrial fungal strains. Front. Bioeng. Biotechnol. 2018, 6, 133. [Google Scholar] [CrossRef]
- Schuerg, T.; Prahl, J.-P.; Gabriel, R.; Harth, S.; Tachea, F.; Chen, C.-S.; Miller, M.; Masson, F.; Matthew, M.; Brown, S.; et al. Xylose induces cellulase production in Thermoascus aurantiacus. Biotechnol. Biofuels 2017, 10, 1–11. [Google Scholar] [CrossRef]
- Granström, T. Metabolic flux analysis of candida tropicalis growing on xylose in an oxygen-limited chemostat. Metab. Eng. 2002, 4, 248–256. [Google Scholar] [CrossRef]
- Hou, J.; Scalcinati, G.; Oldiges, M.; Vemuri, G.N. Metabolic impact of increased NADH availability in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2010, 76, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Christensen, K.E.; MacKenzie, R.E. Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. Bioessays 2006, 28, 595–605. [Google Scholar] [CrossRef]
- Bacher, A.D.; Eberhardt, S.; Fischer, M.; Kis, K.; Richter, G. Biosynthesis of vitamin B2 (riboflavin). Annu. Rev. Nutr. 2000, 20, 153–167. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G., Jr.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, S.; Schüller, H.-J. Transcriptional control of the yeast acetyl-CoA synthetase gene, ACS1, by the positive regulators CAT8 and ADR1 and the pleiotropic repressor UME6. Mol. Microbiol. 1997, 26, 631–641. [Google Scholar] [CrossRef]
- Matsushika, A.; Goshima, T.; Hoshino, T. Transcription analysis of recombinant industrial and laboratory Saccharomyces cerevisiae strains reveals the molecular basis for fermentation of glucose and xylose. Microb. Cell Factories 2014, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, J.; Yuwen, M.; Qian, W.; Wu, X. Yeast-based biosynthesis of natural products from xylose. Front. Bioeng. Biotechnol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Atzmüller, D.; Ullmann, N.; Zwirzitz, A. Identification of genes involved in xylose metabolism of Meyerozyma guilliermondii and their genetic engineering for increased xylitol production. AMB Express 2020, 10, 78. [Google Scholar] [CrossRef]
- Glenn, K.; Ingram-Smith, C.; Smith, K.S. Biochemical and kinetic characterization of xylulose 5-phosphate/fructose 6-phosphate phosphoketolase 2 (Xfp2) from Cryptococcus neoformans. Eukaryot. Cell 2014, 13, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Bergman, A.; Hellgren, J.; Moritz, T.; Siewers, V.; Nielsen, J.; Chen, Y. Heterologous phosphoketolase expression redirects flux towards acetate, perturbs sugar phosphate pools and increases respiratory demand in Saccharomyces cerevisiae. Microb. Cell Factories 2019, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.; Jin, Y.-S. Production of fuels and chemicals from xylose by engineered Saccharomyces cerevisiae: A review and perspective. Microb. Cell Factories 2017, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Pontes, M.V.A.; Brandl, J.; McDonnell, E.; Strasser, K.; Nguyen, T.; Riley, R.; Mondo, S.; Salamov, A.; Nybo, J.; Vesth, T.C.; et al. The gold-standard genome of Aspergillus niger NRRL 3 enables a detailed view of the diversity of sugar catabolism in fungi. Stud. Mycol. 2018, 91, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Toivari, M.H.; Nygård, Y.; Penttilä, M.; Ruohonen, L.; Wiebe, M. Microbial D-xylonate production. Appl. Microbiol. Biotechnol. 2012, 96, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wasserstrom, L.; Portugal-Nunes, D.; Almqvist, H.; Sandström, A.G.; Lidén, G.; Gorwa-Grauslund, M.F. Exploring D-xylose oxidation in Saccharomyces cerevisiae through the Weimberg pathway. AMB Express 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bañares, A.B.; Nisola, G.M.; Valdehuesa, K.N.G.; Lee, W.-K.; Chung, W.-J. Understanding D-xylonic acid accumulation: A cornerstone for better metabolic engineering approaches. Appl. Microbiol. Biotechnol. 2021, 105, 5309–5324. [Google Scholar] [CrossRef]
- Mishra, P.; Park, G.-Y.; Lakshmanan, M.; Lee, H.-S.; Lee, H.; Chang, M.W.; Ching, C.B.; Ahn, J.; Lee, D.-Y. Genome-scale metabolic modeling and in silico analysis of lipid accumulating yeast Candida tropicalis for dicarboxylic acid production. Biotechnol. Bioeng. 2016, 113, 1993–2004. [Google Scholar] [CrossRef]
- Liu, Z.; Gao, Y.; Chen, J.; Imanaka, T.; Bao, J.; Hua, Q. Analysis of metabolic fluxes for better understanding of mechanisms related to lipid accumulation in oleaginous yeast Trichosporon cutaneum. Bioresour. Technol. 2013, 130, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Tehlivets, O.; Scheuringer, K.; Kohlwein, S.D. Fatty acid synthesis and elongation in yeast. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2007, 1771, 255–270. [Google Scholar] [CrossRef]
- Adrio, J.L. Oleaginous yeasts: Promising platforms for the production of oleochemicals and biofuels. Biotechnol. Bioeng. 2017, 114, 1915–1920. [Google Scholar] [CrossRef]
- Ratledge, C.; Wynn, J.P. The biochemistry and molecular biology of lipid accumulation in oleaginous microorganisms. Int. Rev. Cytol. 2002, 51, 1–52. [Google Scholar] [CrossRef]
- Wynn, J.P.; Ratledge, C.; Hamid, A.A.; Li, Y. Biochemical events leading to the diversion of carbon into storage lipids in the oleaginous fungi Mucor circinelloides and Mortierella alpina. Microbiology 2001, 147, 2857–2864. [Google Scholar] [CrossRef] [Green Version]
- Kourist, R.; Bracharz, F.; Lorenzen, J.; Kracht, O.N.; Chovatia, M.; Daum, C.; Deshpande, S.; Lipzen, A.; Nolan, M.; Ohm, R.A.; et al. Genomics and transcriptomics analyses of the oil-accumulating basidiomycete yeast Trichosporon oleaginosus: Insights into substrate utilization and alternative evolutionary trajectories of fungal mating systems. mBio 2015, 6, e00918-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: MTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps, K.R.; Charette, J.M.; Baserga, S.J. The small subunit processome in ribosome biogenesis-progress and prospects. Wiley Interdiscip. Rev. RNA 2010, 2, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Strunk, B.S.; Karbstein, K. Powering through ribosome assembly. RNA 2009, 15, 2083–2104. [Google Scholar] [CrossRef] [Green Version]
- Tiukova, I.A.; Brandenburg, J.; Blomqvist, J.; Sampels, S.; Mikkelsen, N.; Skaugen, M.; Arntzen, M.Ø.; Nielsen, J.; Sandgren, M.; Kerkhoven, E.J. Proteome analysis of xylose metabolism in Rhodotorula toruloides during lipid production. Biotechnol. Biofuels 2019, 12, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, S.D.; Hanley, S.E.; Beishke, T.; Tati, P.D.; Cooper, K.F. Ubiquitin–proteasome-mediated cyclin C degradation promotes cell survival following nitrogen starvation. Mol. Biol. Cell 2020, 31, 1015–1031. [Google Scholar] [CrossRef]
- Pomraning, K.R.; Kim, Y.-M.; Nicora, C.D.; Chu, R.K.; Bredeweg, E.L.; Purvine, S.O.; Hu, D.; Metz, T.O.; Baker, S.E. Multi-omics analysis reveals regulators of the response to nitrogen limitation in Yarrowia lipolytica. BMC Genom. 2016, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Kerkhoven, E.J.; Kim, Y.-M.; Wei, S.; Nicora, C.D.; Fillmore, T.L.; Purvine, S.O.; Webb-Robertson, B.-J.; Smith, R.D.; Baker, S.E.; Metz, T.O.; et al. Leucine biosynthesis is involved in regulating high lipid accumulation in Yarrowia lipolytica. mBio 2017, 8, e00857-17. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Watanabe, R.; Harris, C.L.; Hong, Y.; Ohishi, K.; Kinoshita, K.; Kinoshita, T. PIG-M transfers the first mannose to glycosylphosphatidylinositol on the lumenal side of the ER. EMBO J. 2001, 20, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Sharma, C.B.; Knauer, R.; Lehle, L. Biosynthesis of lipid-linked oligosaccharides in yeast: The ALG3 gene encodes the Dol-P-Man: Man5GlcNAc2-PP-Dol mannosyltransferase. Biol. Chem. 2001, 382, 321–328. [Google Scholar] [CrossRef]
- Bloch, J.; Pesciullesi, G.; Boilevin, J.; Nosol, K.; Irobalieva, R.N.; Darbre, T.; Aebi, M.; Kossiakoff, A.A.; Reymond, J.-L.; Locher, K.P. Structure and mechanism of the ER-based glucosyltransferase ALG6. Nat. Cell Biol. 2020, 579, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Vainauskas, S.; Menon, A. A conserved proline in the last transmembrane segment of Gaa1 Is required for glycosylphosphatidylinositol (GPI) recognition by GPI transamidase. J. Biol. Chem. 2004, 279, 6540–6545. [Google Scholar] [CrossRef] [Green Version]
- Parlati, F.; McNew, J.A.; Fukuda, R.; Miller, R.; Söllner, T.H.; Rothman, J.E. Topological restriction of SNARE-dependent membrane fusion. Nat. Cell Biol. 2000, 407, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, S.; Ben-Tekaya, H.; Koegler, E.; Gruenberg, J.; Hauri, H.-P. The cargo receptors Surf4, endoplasmic reticulum-Golgi intermediate compartment (ERGIC)-53, and p25 are required to maintain the architecture of ERGIC and Golgi. Mol. Biol. Cell 2008, 19, 1976–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
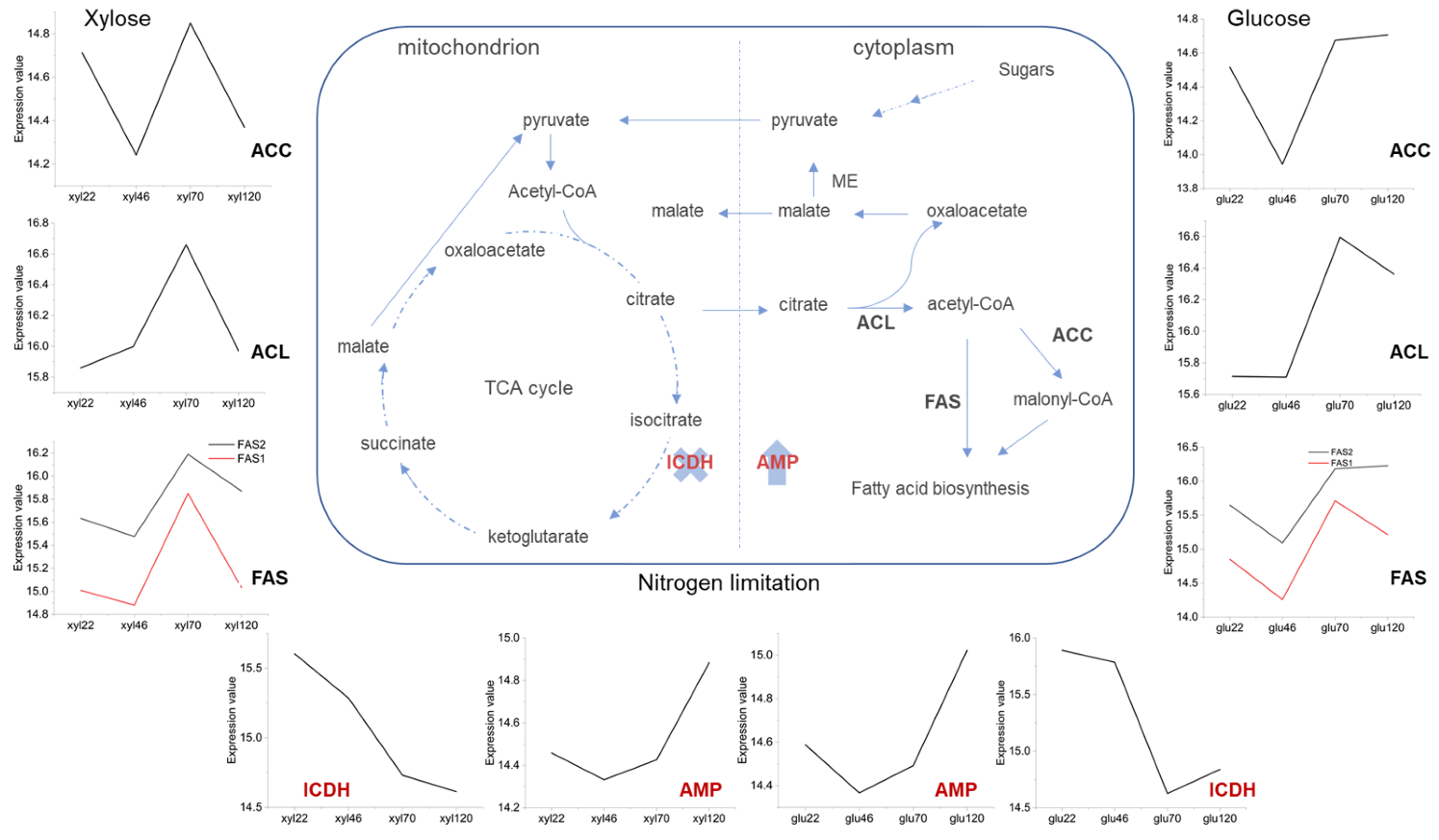{kind=link}
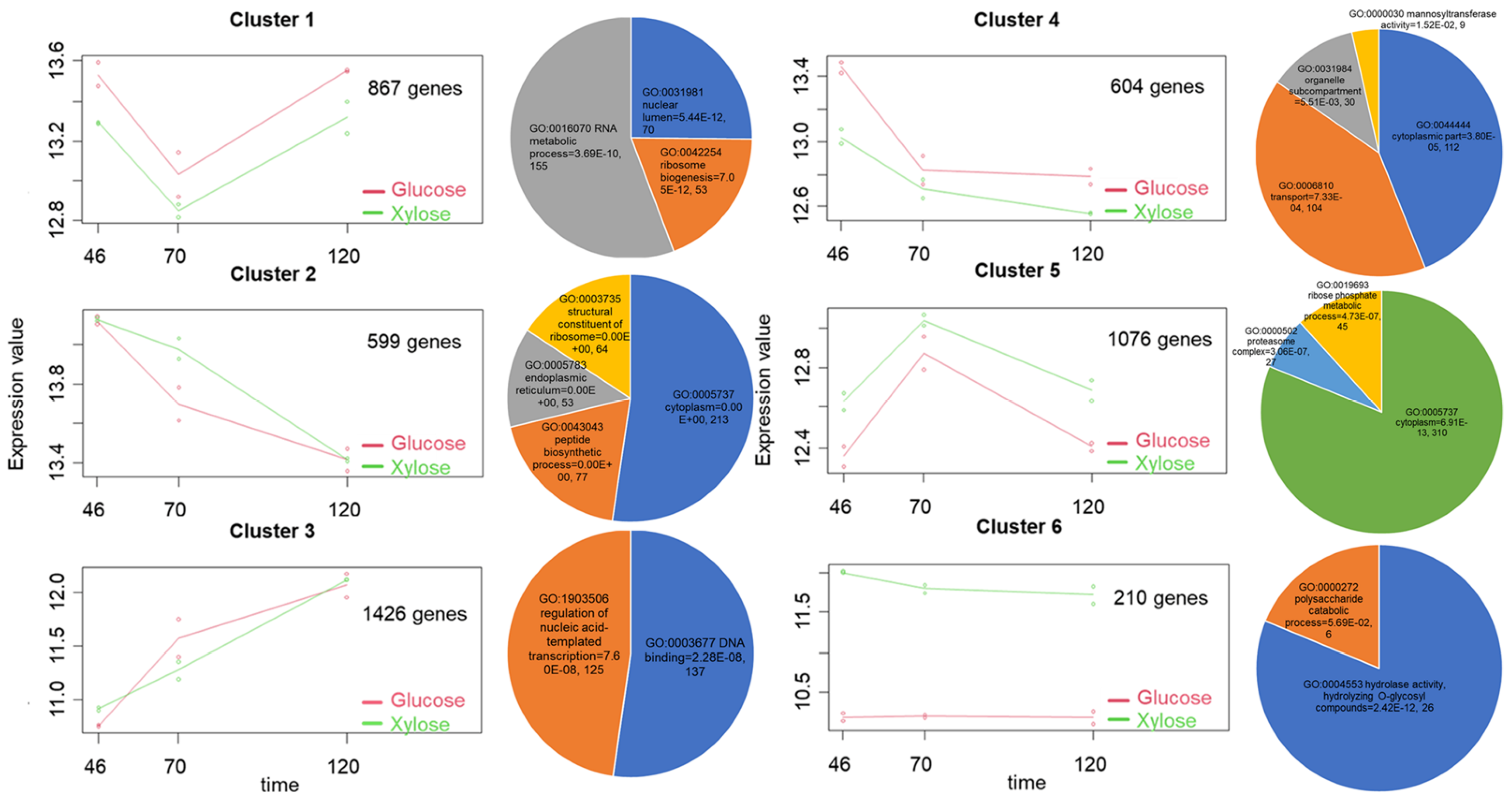{kind=link}
| DE | Category | GO | Ontology Definition | Size | Overlap | Expect | Ratio | p-Value | FDR |
|---|---|---|---|---|---|---|---|---|---|
| Up | BP | GO:0055085 | transmembrane transport | 725 | 15 | 5.08 | 2.95 | 4.4 × 10−5 | 7.1 × 10−2 |
| MF | GO:0022857 | transmembrane transporter activity | 650 | 14 | 4.55 | 3.07 | 5.8 × 10−5 | 7.1 × 10−2 | |
| MF | GO:0005215 | transporter activity | 660 | 14 | 4.62 | 3.03 | 6.9 × 10−5 | 7.1 × 10−2 | |
| Down | MF | GO:0022857 | transmembrane transporter activity | 650 | 51 | 20.69 | 2.46 | 1.8 × 10−10 | 4.7 × 10−7 |
| MF | GO:0005215 | transporter activity | 660 | 51 | 21.01 | 2.43 | 3.1 × 10−10 | 4.7 × 10−7 | |
| MF | GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | 171 | 24 | 5.44 | 4.41 | 4.5 × 10−10 | 4.7 × 10−7 | |
| BP | GO:0055085 | transmembrane transport | 725 | 53 | 23.08 | 2.30 | 9.7 × 10−10 | 7.5 × 10−7 | |
| MF | GO:0016798 | hydrolase activity, acting on glycosyl bonds | 184 | 24 | 5.86 | 4.10 | 2.1 × 10−9 | 1.3 × 10−6 | |
| BP | GO:0005975 | carbohydrate metabolic process | 357 | 33 | 11.36 | 2.90 | 1.3 × 10−8 | 6.7 × 10−6 | |
| BP | GO:0016052 | carbohydrate catabolic process | 89 | 11 | 2.83 | 3.88 | 1.0 × 10−4 | 4.5 × 10−2 | |
| BP | GO:0000272 | polysaccharide catabolic process | 37 | 7 | 1.18 | 5.94 | 1.3 × 10−4 | 5.2 × 10−2 | |
| MF | GO:0004181 | metallocarboxypeptidase activity | 10 | 4 | 0.32 | 12.57 | 1.8 × 10−4 | 6.1 × 10−2 |
| Comparison | DE | Function | GO | Definition | Size | Overlap | Expect | Ratio | p-Value | FDR |
|---|---|---|---|---|---|---|---|---|---|---|
| Glu-Xyl-46 | Up | MF | GO:0022857 | transmembrane transporter activity | 685 | 50 | 26.07 | 1.92 | 2.1 × 10−6 | 5.0 × 10−3 |
| MF | GO:0046915 | transition metal ion transmembrane transporter activity | 21 | 7 | 0.80 | 8.76 | 7.7 × 10−6 | 7.0 × 10−3 | ||
| Down | MF | GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | 179 | 40 | 13.21 | 3.03 | 8.2 × 10−11 | 2.6 × 10−7 | |
| MF | GO:0022857 | transmembrane transporter activity | 685 | 87 | 50.53 | 1.72 | 7.1 × 10−8 | 4.4 × 10−5 | ||
| BP | GO:0045333 | cellular respiration | 48 | 13 | 3.54 | 3.67 | 2.8 × 10−5 | 7.4 × 10−3 | ||
| Glu-Xyl-70 | Down | MF | GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | 179 | 28 | 4.65 | 6.03 | 4.0 × 10−15 | 1.2 × 10−11 |
| BP | GO:0010383 | cell wall polysaccharide metabolic process | 26 | 6 | 0.67 | 8.89 | 4.1 × 10−5 | 2.6 × 10−2 | ||
| BP | GO:0044262 | cellular carbohydrate metabolic process | 106 | 11 | 2.75 | 4.00 | 8.1 × 10−5 | 3.6 × 10−2 | ||
| Glu-Xyl-120 | Down | MF | GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | 179 | 21 | 4.54 | 4.62 | 2.6 × 10−9 | 8.0 × 10−6 |
| Glu-46-70 | Up | CC | GO:0044446 | intracellular organelle part | 1187 | 220 | 152.43 | 1.44 | 7.3 × 10−11 | 2.3 × 10−7 |
| BP | GO:0006364 | rRNA processing | 136 | 43 | 17.46 | 2.46 | 5.6 × 10−9 | 5.8 × 10−6 | ||
| Down | BP | GO:0019752 | carboxylic acid metabolic process | 371 | 115 | 70.45 | 1.63 | 5.3 × 10−9 | 8.7 × 10−6 | |
| BP | GO:0046395 | carboxylic acid catabolic process | 83 | 36 | 15.76 | 2.28 | 2.4 × 10−7 | 1.2 × 10−4 | ||
| BP | GO:1901565 | organonitrogen compound catabolic process | 230 | 75 | 43.68 | 1.72 | 3.3 × 10−7 | 1.3 × 10−4 | ||
| Glu-70-120 | Up | CC | GO:0044444 | cytoplasmic part | 1220 | 260 | 156.19 | 1.66 | 0 | 0 |
| BP | GO:1901564 | organonitrogen compound metabolic process | 1486 | 279 | 190.25 | 1.47 | 1.6 × 10−15 | 1.6 × 10−12 | ||
| CC | GO:0044429 | mitochondrial part | 205 | 67 | 26.25 | 2.55 | 3.2 × 10−14 | 2.0 × 10−11 | ||
| Down | CC | GO:0005634 | nucleus | 1293 | 217 | 135.76 | 1.60 | 2.2 × 10−16 | 6.9 × 10−13 | |
| BP | GO:0051171 | regulation of nitrogen compound metabolic process | 665 | 113 | 69.82 | 1.62 | 2.5 × 10−8 | 1.1 × 10−5 | ||
| Xyl-46-70 | Up | CC | GO:1990904 | ribonucleoprotein complex | 380 | 76 | 27.29 | 2.78 | 0 | 0 |
| BP | GO:0042254 | ribosome biogenesis | 189 | 60 | 13.57 | 4.42 | 0 | 0 | ||
| BP | GO:0016072 | rRNA metabolic process | 153 | 44 | 10.99 | 4.00 | 2.2 × 10−16 | 8.6 × 10−14 | ||
| Down | BP | GO:1901565 | organonitrogen compound catabolic process | 230 | 41 | 19.57 | 2.10 | 2.8 × 10−6 | 8.8 × 10−3 | |
| MF | GO:0048038 | quinone binding | 13 | 7 | 1.11 | 6.33 | 3.4 × 10−5 | 3.1 × 10−2 | ||
| BP | GO:0006637 | acyl-CoA metabolic process | 23 | 9 | 1.96 | 4.60 | 6.0 × 10−5 | 3.1 × 10−2 | ||
| Xyl-70-120 | Up | CC | GO:0005737 | cytoplasm | 1677 | 296 | 198.99 | 1.49 | 0 | 0 |
| BP | GO:0009150 | purine ribonucleotide metabolic process | 106 | 39 | 12.58 | 3.10 | 1.7 × 10−11 | 1.8 × 10−8 | ||
| Down | BP | GO:0042254 | ribosome biogenesis | 189 | 53 | 20.88 | 2.54 | 3.9 × 10−11 | 4.0 × 10−8 | |
| BP | GO:0016070 | RNA metabolic process | 1029 | 173 | 113.66 | 1.52 | 2.0 × 10−10 | 1.5 × 10−7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliyu, H.; Gorte, O.; Neumann, A.; Ochsenreither, K. Global Transcriptome Profile of the Oleaginous Yeast Saitozyma podzolica DSM 27192 Cultivated in Glucose and Xylose. J. Fungi 2021, 7, 758. https://doi.org/10.3390/jof7090758
Aliyu H, Gorte O, Neumann A, Ochsenreither K. Global Transcriptome Profile of the Oleaginous Yeast Saitozyma podzolica DSM 27192 Cultivated in Glucose and Xylose. Journal of Fungi. 2021; 7(9):758. https://doi.org/10.3390/jof7090758
Chicago/Turabian StyleAliyu, Habibu, Olga Gorte, Anke Neumann, and Katrin Ochsenreither. 2021. "Global Transcriptome Profile of the Oleaginous Yeast Saitozyma podzolica DSM 27192 Cultivated in Glucose and Xylose" Journal of Fungi 7, no. 9: 758. https://doi.org/10.3390/jof7090758