
The Abundance and Diversity of Fungi in a Hypersaline Microbial Mat from Guerrero Negro, Baja California, México



Abstract
:1. Introduction
2. Materials and Methods
2.1. Microbial Mat Samples Collection
2.2. Oxygen Profiles and Microscope Analysis
2.3. DNA Extraction and qPCR Analysis
2.4. Metagenomic Sequencing
2.5. Metagenomic Processing
2.6. Statistical Analysis
3. Results
3.1. Microscope Analysis and Oxygen Profiles
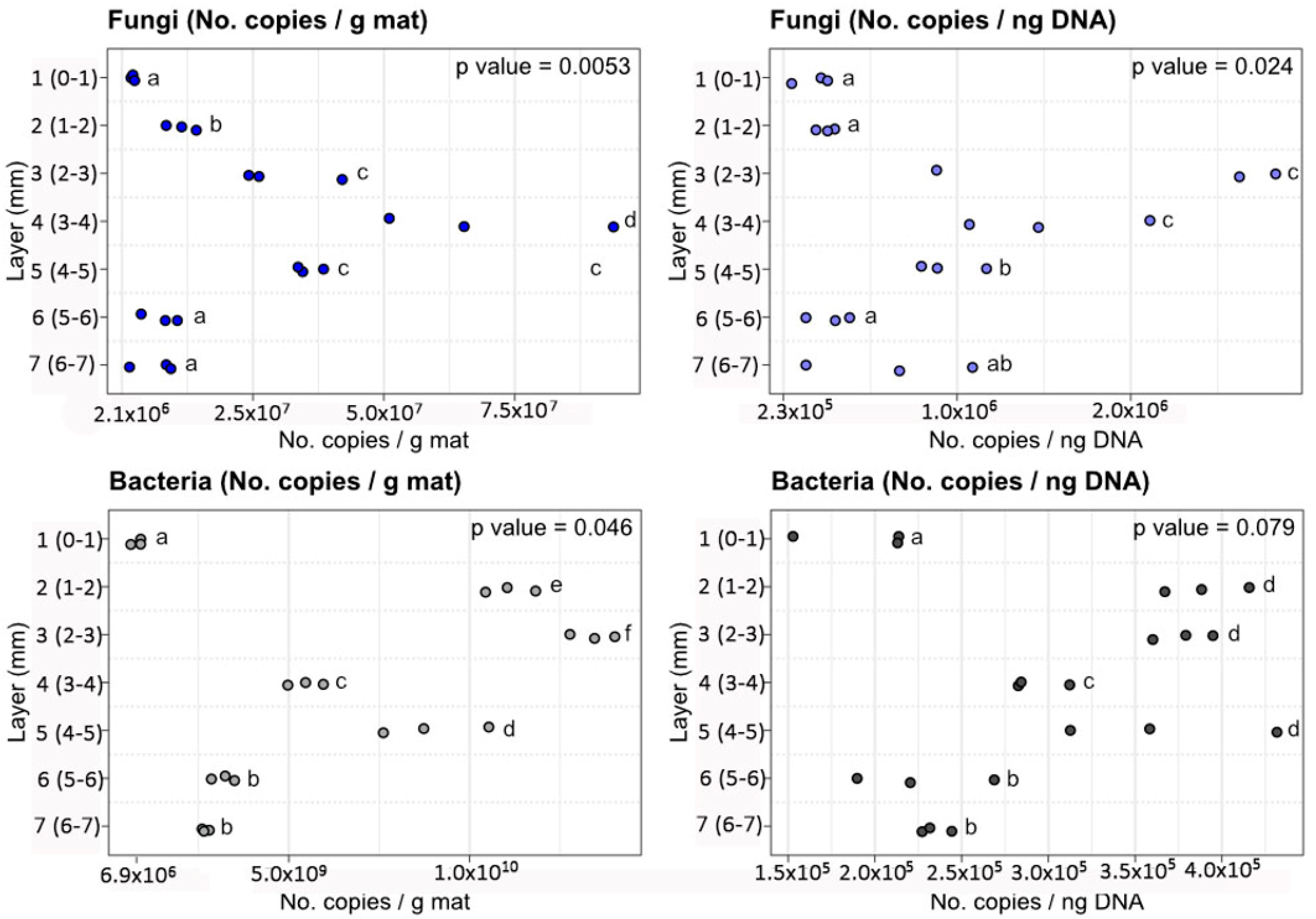
3.2. Abundance of Fungal and Bacterial Derived Sequenced as Determined by qPCR
3.3. Metagenomics
4. Discussion
4.1. Abundance of Fungi in the Guerrero Negro Microbial Mat
4.2. Diversity of Fungi in the Guerrero Negro Microbial Mat
4.3. Potential Roles of Fungi in the Guerrero Negro Microbial Mat
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stal, L.J.; Noffke, N. Microbial Mats. In Encyclopedia of Astrobiology; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J., Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1042–1045. [Google Scholar]
- Hoehler, T.M.; Bebout, B.M.; Des Marais, D.J. The Role of Microbial Mats in the Production of Reduced Gases on the Early Earth. Nature 2001, 412, 324–327. [Google Scholar] [CrossRef]
- Fenchel, T.; Blackburn, H.; King, G.M.; Blackburn, T.H. Bacterial Biogeochemistry: The Ecophysiology of Mineral Cycling; Academic Press: Cambridge, MA, USA, 2012. [Google Scholar]
- White, R.A., III.; Wong, H.L.; Ruvindy, R.; Neilan, B.A.; Burns, B.P. Viral Communities of Shark Bay Modern Stromatolites. Front. Microbiol. 2018, 9, 1223. [Google Scholar] [CrossRef] [Green Version]
- Stal, L.J.; van Gemerden, H.; Krumbein, W.E. Structure and Development of a Benthic Marine Microbial Mat. FEMS Microbiol. Ecol. 1985, 111–125. [Google Scholar] [CrossRef]
- Stal, L.J. Cyanobacterial Mats and Stromatolites. In Ecology of Cyanobacteria Ii: Their Diversity in Space and Time; Whitton, B.A., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 65–125. [Google Scholar]
- Ley, R.; Harris, J.; Wilcox, J.; Spear, J.; Miller, S.; Bebout, B.; Maresca, J.; Bryant, D.; Sogin, M.; Pace, N. Unexpected Diversity and Complexity of the Guerrero Negro Hypersaline Microbial Mat. Appl. Environ. Microbiol. 2006, 72, 3685–3695. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.L.; Smith, D.-L.; Visscher, P.T.; Burns, B.P. Niche Differentiation of Bacterial Communities at a Millimeter Scale in Shark Bay Microbial Mats. Sci. Rep. 2015, 5, 15607. [Google Scholar] [CrossRef]
- Carreira, C.; Lønborg, C.; Kühl, M.; Lillebø, A.I.; Sandaa, R.-A.; Villanueva, L.; Cruz, S. Fungi and Viruses as Important Players in Microbial Mats. FEMS Microbiol. Ecol. 2020, 96, 11. [Google Scholar] [CrossRef] [PubMed]
- Giani, D.; Seeler, J.; Giani, L.; Krumbein, W.E. Microbial Mats and Physicochemistry in a Saltern in the Bretagne (France) and in a Laboratory Scale Saltern Model. FEMS Microbiol. Ecol. 1989, 5, 151–161. [Google Scholar] [CrossRef]
- Cantrell, S.A.; Casillas-Martínez, L.; Molina, M. Characterization of Fungi from Hypersaline Environments of Solar Salterns Using Morphological and Molecular Techniques. Mycol. Res. 2006, 110, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Panzer, K.; Glöckner, F.O.; Reich, M.A. Phylogenetic Framework for the Kingdom Fungi Based on 18s Rrna Gene Sequences. Mar. Genomics 2017, 36, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Maza-Márquez, P.; Vílchez-Vargas, R.; González-Martínez, A.; González-López, J.; Rodelas, B. Assessing the Abundance of Fungal Populations in a Full-Scale Membrane Bioreactor (Mbr) Treating Urban Wastewater by Using Quantitative PCR (qPCR). J. Environ. Manag. 2018, 223, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear Ribosomal Internal Transcribed Spacer (Its) Region as a Universal DNA Barcode Marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebout, B.; Carpenter, S.; Des Marais, D.; Discipulo, M.; Embaye, T.; Garcia-Pichel, F.; Hoehler, T.; Hogan, M.; Jahnke, L.; Keller, R.; et al. Long-Term Manipulations of Intact Microbial Mat Communities in a Greenhouse Collaboratory: Simulating Earth’s Present and Past Field Environments. Astrobiology 2002, 2, 383–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coban, O.; Williams, M.; Bebout, B.M. Mechanisms of Nitrogen Attenuation from Seawater by Two Microbial Mats. Water Res. 2018, 147, 373–381. [Google Scholar] [CrossRef]
- Omoregie, E.O.; Crumbliss, L.L.; Bebout, B.M.; Zehr, J.P. Comparison of Diazotroph Community Structure in Lyngbya Sp. And Microcoleus Chthonoplastes Dominated Microbial Mats from Guerrero Negro, Baja, Mexico. FEMS Microbiol. Ecol. 2004, 47, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16s Rrna. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.M.; Kachur, S.; Dwan, M.G.; Abraham, A.G.; Aziz, M.; Hsueh, P.R.; Huang, Y.T.; Busch, J.D.; Lamit, L.J.; Gehring, C.A.; et al. Fungiquant: A Broad-Coverage Fungal Quantitative Real-Time PCR Assay. BMC Microbiol. 2012, 12, 1471–2180. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. Fastqc. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 March 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating Species Abundance in Metagenomics Data. Peer J Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. Past: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. Open J. Stat. 2017, 7, 5. [Google Scholar]
- Buchalo, A.S.; Nevo, E.; Wasser, S.P.; Oren, A.; Molitoris, H.P. Fungal Life in the Extremely Hypersaline Water of the Dead Sea: First Records. Proc. Biol. Sci. 1998, 265, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Kis-Papo, T.; Oren, A.; Wasser, S.P.; Nevo, E. Survival of Filamentous Fungi in Hypersaline Dead Sea Water. Microbiol. Ecol. 2003, 45, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Gunde-Cimerman, N.; Zalar, P.; Petrovič, U.; Turk, M.; Kogej, T.; de Hoog, G.S.; Plemenitaš, A. Fungi in Salterns. In Halophilic Microorganisms; Ventosa, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 103–113. [Google Scholar]
- Butinar, L.; Sonjak, S.; Zalar, P.; Plemenitaš, A.; Gunde-Cimerman, N. Melanized Halophilic Fungi Are Eukaryotic Members of Microbial Communities in Hypersaline Waters of Solar Salterns. Bot. Mar. 2005, 48, 73–79. [Google Scholar] [CrossRef]
- Feazel, L.M.; Spear, J.R.; Berger, A.B.; Harris, J.K.; Frank, D.N.; Ley, R.E.; Pace, N.R. Eucaryotic Diversity in a Hypersaline Microbial Mat. Appl. Environ. Microbiol. 2008, 74, 329–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.A.; Goh, F.; Burns, B.P.; Neilan, B.A. Bacterial, Archaeal and Eukaryotic Diversity of Smooth and Pustular Microbial Mat Communities in the Hypersaline Lagoon of Shark Bay. Geobiology 2009, 7, 82–96. [Google Scholar] [CrossRef]
- Maza-Márquez, P.; Castellano-Hinojosa, A.; González-Martínez, A.; Juárez-Jiménez, B.; González-López, J.; Rodelas, B. Abundance of Total and Metabolically Active Candidatus Microthrix and Fungal Populations in Three Full-Scale Wastewater Treatment Plants. Chemosphere 2019, 232, 26–34. [Google Scholar] [CrossRef]
- Gallardo-Altamirano, M.J.; Maza-Márquez, P.; Peña-Herrera, J.M.; Rodelas, B.; Osorio, F.; Pozo, C. Removal of Anti-Inflammatory/Analgesic Pharmaceuticals from Urban Wastewater in a Pilot-Scale a(2)O System: Linking Performance and Microbial Population Dynamics to Operating Variables. Sci. Total Environ. 2018, 643, 1481–1492. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, G.; Torralbo, F.; Maza-Márquez, P.; Aranda, E.; Calvo, C.; González-Murua, C.; Bedmar, E.J. Assessment of the Diversity and Abundance of the Total and Active Fungal Population and Its Correlation with Humification During Two-Phase Olive Mill Waste (‘‘Alperujo) Composting. Bioresour. Technol. 2020, 295, 122267. [Google Scholar] [CrossRef]
- Maza-Márquez, P.; Vilchez-Vargas, R.; Kerckhof, F.M.; Aranda, E.; González-López, J.; Rodelas, B. Community Structure, Population Dynamics and Diversity of Fungi in a Full-Scale Membrane Bioreactor (Mbr) for Urban Wastewater Treatment. Water Res. 2016, 105, 507–519. [Google Scholar] [CrossRef]
- Cheung, M.K.; Wong, C.K.; Chu, K.H.; Kwan, H.S. Community Structure, Dynamics and Interactions of Bacteria, Archaea and Fungi in Subtropical Coastal Wetland Sediments. Sci. Rep. 2018, 8, 14397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, J.P.; González, M.; Cardinale, M.; Schnell, S.; Stoll, A. Microbiome Dynamics Associated with the Atacama Flowering Desert. Front. Microbiol. 2020, 10, 3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogwu, M.C.; Takahashi, K.; Dong, K.; Song, H.-K.; Moroenyane, I.; Waldman, B.; Adams, J.M. Fungal Elevational Rapoport Pattern from a High Mountain in Japan. Sci. Rep. 2019, 9, 6570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Jia, X.; Yan, W.; Zhong, Y.; Shangguan, Z. Changes in Soil Microbial Community Structure During Long-Term Secondary Succession. Land Degrad. Dev. 2020, 31, 1151–1166. [Google Scholar] [CrossRef]
- Cantrell, S.; Duval-Pérez, L. Microbial Mats: An Ecological Niche for Fungi. Front. Microbiol. 2013, 3, 424. [Google Scholar] [CrossRef] [Green Version]
- Grossart, H.-P.; Rojas-Jimenez, K. Aquatic Fungi: Targeting the Forgotten in Microbial Ecology. Curr. Opin. Microbiol. 2016, 31, 140–145. [Google Scholar] [CrossRef]
- Aranda, E. Promising Approaches Towards Biotransformation of Polycyclic Aromatic Hydrocarbons with Ascomycota Fungi. Curr. Opin. Biotechnol. 2016, 38, 1–8. [Google Scholar] [CrossRef]
- Keeling, P.J.; Fast, N.M. Microsporidia: Biology and Evolution of Highly Reduced Intracellular Parasites. Annu. Rev. Microbiol. 2002, 56, 93–116. [Google Scholar] [CrossRef] [Green Version]
- Varin, T.; Lovejoy, C.; Jungblut, A.D.; Vincent, W.F.; Corbeil, J. Metagenomic Profiling of Arctic Microbial Mat Communities as Nutrient Scavenging and Recycling Systems. Limnol. Oceanogr. 2010, 55, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.R.; Tudryn, G.; Bucinell, R.; Schadler, L.; Picu, R.C. Morphology and Mechanics of Fungal Mycelium. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Malkin, S.Y.; Meysman, F.J.R. Rapid Redox Signal Transmission by Cable Bacteria beneath a Photosynthetic Biofilm. Appl. Environ. Microbiol. 2015, 81, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, S.D.; Hofmann, A.; Pilhofer, M.; Wanner, G.; Agerer, R.; Ludwig, W.; Schleifer, K.H.; Fried, J. The Diversity of Fungi in Aerobic Sewage Granules Assessed by 18s Rrna Gene and Its Sequence Analyses. FEMS Microbiol. Ecol. 2009, 68, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.O.; Blackwell, M.; Kurtzman, C.P.; Lachance, M.A. Phylogenetics of Saccharomycetales, the Ascomycete Yeasts. Mycologia 2006, 98, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Dighton, J. Nutrient Cycling by Saprotrophic Fungi in Terrestrial Habitats. In Environmental and Microbial Relationships; Kubicek, C.P., Druzhinina, I.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 287–300. [Google Scholar]
- Mahmood, A.; Iguchi, R.; Kataoka, R. Multifunctional Food Waste Fertilizer Having the Capability of Fusarium-Growth Inhibition and Phosphate Solubility: A New Horizon of Food Waste Recycle Using Microorganisms. Waste Manag. 2019, 94, 77–84. [Google Scholar] [CrossRef]
- Kagami, M.; Miki, T.; Takimoto, G. Mycoloop: Chytrids in Aquatic Food Webs. Front. Microbiol. 2014, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Moretto, M.; Weiss, L.M. Encephalitozoon: Tissue Culture, Cryopreservation, and Murine Infection. Curr. Protoc. Microbiol. 2019, 52, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreira, C.; Staal, M.; Falkoski, D.; de Vries, R.P.; Middelboe, M.; Brussaard, C.P. Disruption of Photoautotrophic Intertidal Mats by Filamentous Fungi. Environ. Microbiol. 2015, 17, 2910–2921. [Google Scholar] [CrossRef]
- Cathrine, S.J.; Raghukumar, C. Anaerobic Denitrification in Fungi from the Coastal Marine Sediments Off Goa, India. Mycol. Res. 2009, 113, 100–109. [Google Scholar] [CrossRef]
- Zhou, Z.; Takaya, N.; Sakairi, M.A.C.; Shoun, H. Oxygen Requirement for Denitrification by the Fungus Fusarium Oxysporum. Arch. Microbiol. 2001, 175, 19–25. [Google Scholar] [CrossRef]
- Morozkina, E.V.; Kurakov, A.V. Dissimilatory Nitrate Reduction in Fungi under Conditions of Hypoxia and Anoxia: A Review. Prikl Biokhim Mikrobiol 2007, 43, 607–613. [Google Scholar] [CrossRef]
- Shiro, Y.; Fujii, M.; Isogai, Y.; Adachi, S.-I.; Iizuka, T.; Obayashi, E.; Makino, R.; Nakahara, K.; Shoun, H. Iron-Ligand Structure and Iron Redox Property of Nitric Oxide Reductase Cytochrome P450nor from Fusarium Oxysporum: Relevance to Its No Reduction Activity. Biochemistry 1995, 34, 9052–9058. [Google Scholar] [CrossRef]
- Zumft, W.G. Cell Biology and Molecular Basis of Denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [CrossRef]
- Coban, O.; Rasigraf, O.; de Jong, A.E.E.; Spott, O.; Bebout, B.M. Quantifying Potential N Turnover Rates in Hypersaline Microbial Mats by 15N tracer techniques. Appl. Environ. Microbiol. 2021. [Google Scholar] [CrossRef]
- Rusticucci, M.; Brönnimann, S.; Charabi, Y.; Dentener, F.; Easterling, D.; Soden, B.; Thorne, P.; Wild, M.; Zhai, P.; Allan, R.; et al. Ipcc (2013), Climate Change 2013, in the Physical Science Basis, Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2014; pp. 159–254. [Google Scholar]
- Novinscak, A.; Goyer, C.; Zebarth, B.J.; Burton, D.L.; Chantigny, M.H.; Filion, M. Novel P450nor Gene Detection Assay Used to Characterize the Prevalence and Diversity of Soil Fungal Denitrifiers. Appl. Environ. Microbiol. 2016, 82, 4560–4569. [Google Scholar] [CrossRef] [Green Version]
- Herold, M.; Baggs, L.; Daniell, T. Fungal and Bacterial Denitrification Are Differently Affected by Long-Term Ph Amendment and Cultivation of Arable Soil. Soil Biol. Biochem. 2012, 54, 25–35. [Google Scholar] [CrossRef]
- Laughlin, R.J.; Rütting, T.; Müller, C.; Watson, C.J.; Stevens, R.J. Effect of Acetate on Soil Respiration, N2o Emissions and Gross N Transformations Related to Fungi and Bacteria in a Grassland Soil. Appl. Soil Ecol. 2009, 42, 25–30. [Google Scholar] [CrossRef]
- Marusenko, Y.; Huber, D.P.; Hall, S.J. Fungi Mediate Nitrous Oxide Production but Not Ammonia Oxidation in Aridland Soils of the Southwestern Us. Soil Biol. Biochem. 2013, 63, 24–36. [Google Scholar] [CrossRef]
- Baroncelli, R.; Sanz-Martín, J.M.; Rech, G.E.; Sukno, S.A.; Thon, M.R. Draft Genome Sequence of Colletotrichum Sublineola, a Destructive Pathogen of Cultivated Sorghum. Genome Announc. 2014, 2, e00540-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elad, Y.; Vivier, M.; Fillinger, S. Botrytis, the Good, the Bad and the Ugly. In Botrytis—The Fungus, the Pathogen and Its Management in Agricultural Systems; Fillinger, S., Elad, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–15. [Google Scholar]
- Brefort, T.; Doehlemann, G.; Mendoza-Mendoza, A.; Reissmann, S.; Djamei, A.; Kahmann, R. Ustilago Maydis as a Pathogen. Annu. Rev. Phytopathol. 2009, 47, 423–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larriba, E.; Jaime, M.D.; Carbonell-Caballero, J.; Conesa, A.; Dopazo, J.; Nislow, C.; Martín-Nieto, J.; Lopez-Llorca, L.V. Sequencing and Functional Analysis of the Genome of a Nematode Egg-Parasitic Fungus, Pochonia Chlamydosporia. Fungal Genet. Biol. 2014, 65, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askun, T. Introductory Chapter: Fusarium: Pathogenicity, Infections, Diseases, Mycotoxins and Management. In Fusarium: Plant Diseases, Pathogen Diversity, Genetic Diversity, Resistance and Molecular Markers; BoD—Books on Demand: Norderstedt, Germany, 2018; p. 1. [Google Scholar]
- Zeigler, R.S.; Finckh, M.R. Pathogenic Variability of Pyricularia Grisea from the High- and Mid-Elevation Zones of Bhutan. Phytopathology 2000, 90, 621–628. [Google Scholar]
- Hospenthal, D.R.; Rinaldi, M.G. Diagnosis and Treatment of Fungal Infections; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer, (Depth Below Surface) | Oxygen Concentration (µM) |
|---|---|
| Layer 1, (0–1 mm) | 200–800 |
| Layer 2, (1–2 mm) | 0–1200 |
| Layer 3, (2–3 mm) | 0–200 |
| Layer 4, (3–4 mm) | Only detectable under very high light |
| Layers 5–7, (4–7 mm) | not detectable |
| A. Fungi | B. Bacteria | |||
|---|---|---|---|---|
| Layer, (Depth Below Surface) | Copies/g mat | Copies/ng DNA | Copies/g mat | Copies/ng DNA |
| Layer 1, (0–1 mm) | 2.47 × 106 ± 3.42 × 105 | 1.84 × 105 ± 1.10 × 105 | 8.27 × 108 ± 1.93 × 108 | 1.85 × 105 ± 4.31 × 104 |
| Layer 2, (1–2 mm) | 1.17 × 107 ± 2.88 × 106 | 2.57 × 105 ± 5.46 × 104 | 1.12 × 1010 ± 9.80 × 108 | 3.93 × 105 ± 3.44 × 104 |
| Layer 3, (2–3 mm) | 3.12 × 107 ± 9.78 × 106 | 2.13 × 106 ± 1.07 × 106 | 1.35 × 1010 ± 8.76 × 108 | 3.79 × 105 ± 2.47 × 104 |
| Layer 4, (3–4 mm) | 7.05 × 107 ± 2.81 × 107 | 1.56 × 106 ± 5.25 × 105 | 5.35 × 109 ± 4.51 × 108 | 3.02 × 105 ± 2.54 × 104 |
| Layer 5, (4–5 mm) | 3.59 × 107 ± 2.59 × 106 | 9.62 × 105 ± 1.95 × 105 | 9.06 × 109 ± 1.97 × 109 | 3.71 × 105 ± 8.05 × 104 |
| Layer 6, (5–6 mm) | 7.90 × 106 ± 3.55 × 106 | 2.82 × 105 ± 1.28 × 105 | 3.24 × 109 ± 4.50 × 108 | 2.46 × 105 ± 3.42 × 104 |
| Layer 7, (6–7 mm) | 6.80 × 106 ± 4.32 × 106 | 6.41 × 105 ± 4.81 × 105 | 2.76 × 109 ± 1.41 × 108 | 2.37 × 105 ± 1.21 × 104 |
| Phylum | Class | Order | Family | Genus | Average% |
|---|---|---|---|---|---|
| Ascomycota | Dothideomycetes | Capnodiales | Mycosphaerellaceae | Cercospora | 2.20 |
| Zymoseptoria | 2.86 | ||||
| Eurotiomycetes | Eurotiales | Aspergillaceae | Aspergillus | 3.93 | |
| Leotiomycetes | Helotiales | Sclerotiniaceae | Botrytis | 4.39 | |
| Saccharomycetes | Saccharomycetales | Debaryomycetaceae | Candida | 4.62 | |
| Debaryomyces | 0.87 | ||||
| Scheffersomyces | 0.93 | ||||
| Dipodascaceae | Yarrowia | 2.16 | |||
| Phaffomycetaceae | Komagataella | 0.84 | |||
| Pichiaceae | Ogataea | 0.88 | |||
| Pichia | 1.08 | ||||
| Saccharomycetaceae | Eremothecium | 2.11 | |||
| Kazachstania | 1.50 | ||||
| Kluyveromyces | 1.69 | ||||
| Lachancea | 0.88 | ||||
| Nakaseomyces | 3.26 | ||||
| Saccharomyces | 1.77 | ||||
| Tetrapisispora | 2.74 | ||||
| Torulaspora | 0.99 | ||||
| Zygosaccharomyces | 0.64 | ||||
| Trichomonascaceae | Sugiyamaella | 1.14 | |||
| Schizosaccharomycetes | Schizosaccharomycetales | Schizosaccharomycetaceae | Schizosaccharomyces | 0.88 | |
| Sordariomycetes | Sordariales | Chaetomiaceae | Thermothelomyces | 11.14 | |
| Hypocreales | Clavicipitaceae | Pochonia | 2.89 | ||
| Glomerellales | Glomerellaceae | Colletotrichum | 7.00 | ||
| Nectriaceae | Fusarium | 11.68 | |||
| Magnaporthales | Pyriculariaceae | Pyricularia | 10.66 | ||
| Sordariales | Sordariaceae | Neurospora | 4.72 | ||
| Basidiomycota | Tremellomycetes | Tremellales | Cryptococcaceae | Cryptococcus | 2.96 |
| Malasseziomycetes | Malasseziales | Malasseziaceae | Malassezia | 0.92 | |
| Ustilaginomycetes | Ustilaginales | Ustilaginaceae | Sporisorium | 2.27 | |
| Ustilago | 2.94 | ||||
| Microsporidia | NA | NA | Unikaryonidae | Encephalitozoon | 0.45 |
| Av.Abund. | Av.Sim. | Sim./S.D. | Contrib. | Cum. | |
|---|---|---|---|---|---|
| Fusarium | 11.68 | 11.27 | 134.85 | 12.02 | 12.02 |
| Thermothelomyces | 11.14 | 10.58 | 19.09 | 11.29 | 23.31 |
| Pyricularia | 10.66 | 10.43 | 35.85 | 11.13 | 34.44 |
| Colletotrichum | 7 | 6.7 | 18.19 | 7.15 | 41.59 |
| Candida | 4.62 | 4.51 | 48.27 | 4.82 | 46.4 |
| Neurospora | 4.72 | 4.49 | 39.81 | 4.79 | 51.19 |
| Botrytis | 4.39 | 3.91 | 4.82 | 4.17 | 55.36 |
| Aspergillus | 3.94 | 3.72 | 12.63 | 3.97 | 59.33 |
| Nakaseomyces | 3.26 | 3.02 | 17.23 | 3.22 | 62.55 |
| Cryptococcus | 2.96 | 2.83 | 31.55 | 3.02 | 65.57 |
| Pochonia | 2.9 | 2.74 | 19.25 | 2.92 | 68.49 |
| Zymoseptoria | 2.86 | 2.6 | 22.96 | 2.78 | 71.27 |
| Tetrapisispora | 2.74 | 2.47 | 6.21 | 2.64 | 73.91 |
| Ustilago | 2.94 | 2.45 | 6.92 | 2.62 | 76.53 |
| Sporisorium | 2.27 | 2.26 | 274.52 | 2.41 | 78.94 |
| Yarrowia | 2.16 | 2.09 | 41.1 | 2.23 | 81.17 |
| Cercospora | 2.2 | 1.96 | 10.7 | 2.09 | 83.26 |
| Eremothecium | 2.11 | 1.93 | 6.64 | 2.06 | 85.32 |
| Saccharomyces | 1.77 | 1.64 | 20.02 | 1.75 | 87.07 |
| Kluyveromyces | 1.7 | 1.61 | 18.45 | 1.71 | 88.79 |
| Kazachstania | 1.5 | 1.4 | 12.92 | 1.5 | 90.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maza-Márquez, P.; Lee, M.D.; Bebout, B.M. The Abundance and Diversity of Fungi in a Hypersaline Microbial Mat from Guerrero Negro, Baja California, México. J. Fungi 2021, 7, 210. https://doi.org/10.3390/jof7030210
Maza-Márquez P, Lee MD, Bebout BM. The Abundance and Diversity of Fungi in a Hypersaline Microbial Mat from Guerrero Negro, Baja California, México. Journal of Fungi. 2021; 7(3):210. https://doi.org/10.3390/jof7030210
Chicago/Turabian StyleMaza-Márquez, Paula, Michael D. Lee, and Brad M. Bebout. 2021. "The Abundance and Diversity of Fungi in a Hypersaline Microbial Mat from Guerrero Negro, Baja California, México" Journal of Fungi 7, no. 3: 210. https://doi.org/10.3390/jof7030210