
Immunosuppressive Sesquiterpenoids from the Edible Mushroom Craterellus odoratus


Abstract
:1. Introduction
2. Materials and Methods
2.1. General Experimental Procedures
2.2. Fungal Material
2.3. Fermentation, Extraction and Isolation
2.4. NMR and ECD Calculations
2.5. Cytotoxicity Assay
2.6. Immunosuppressive Activities Assay
2.6.1. Preparation of Spleen Cells from Mice
2.6.2. Cytotoxicity Assay
2.6.3. T and B Cell Function Assay
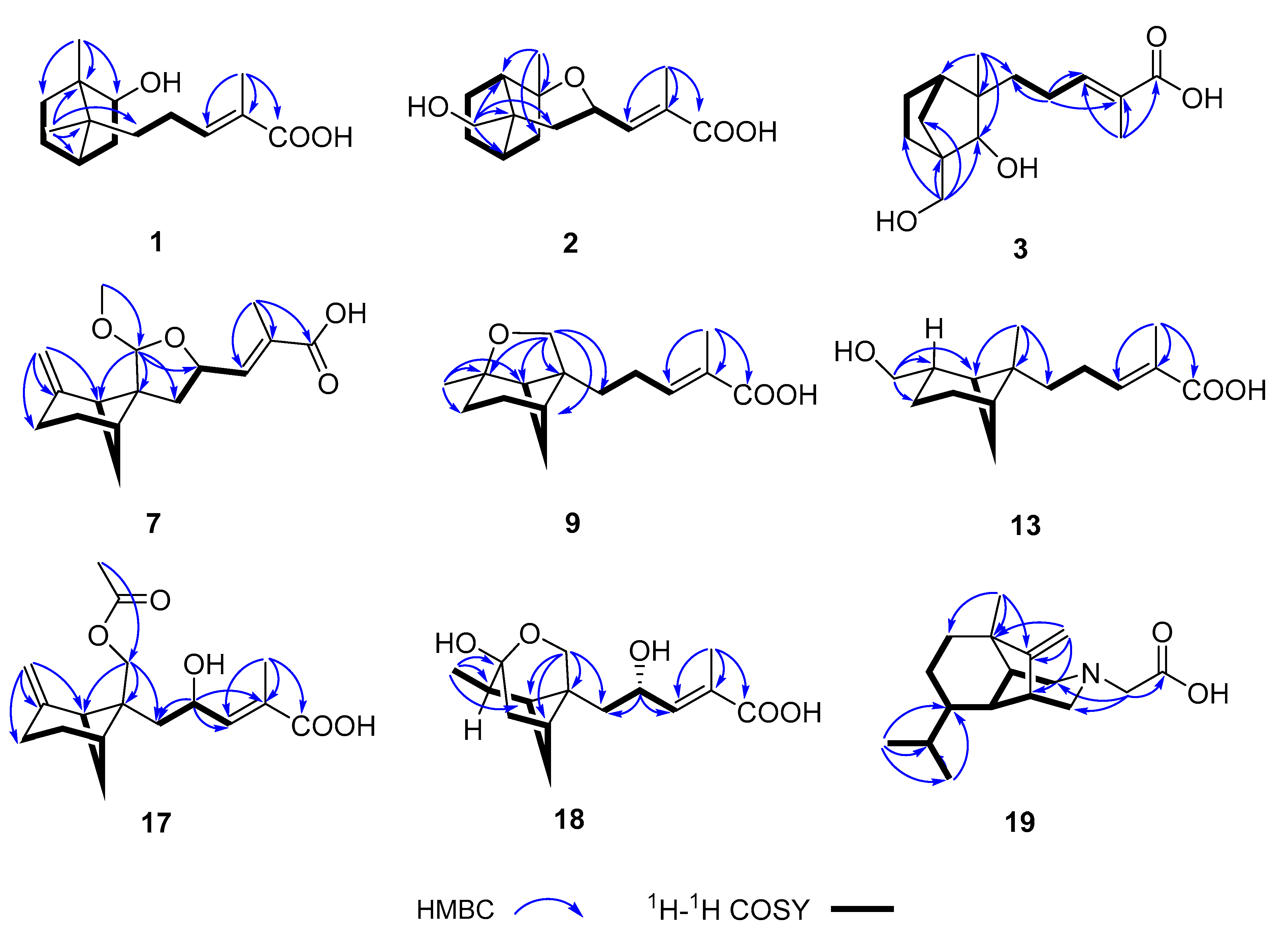
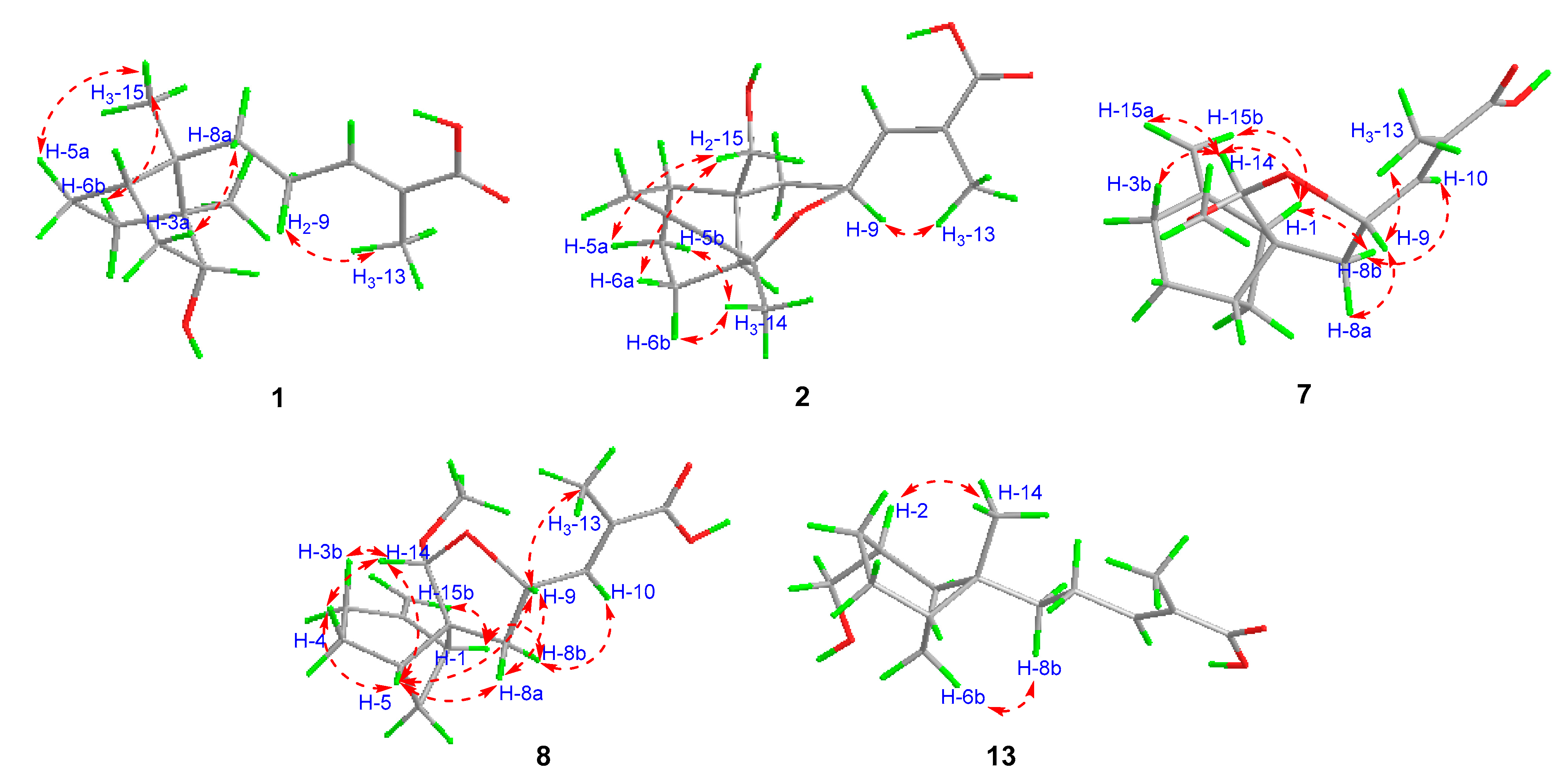
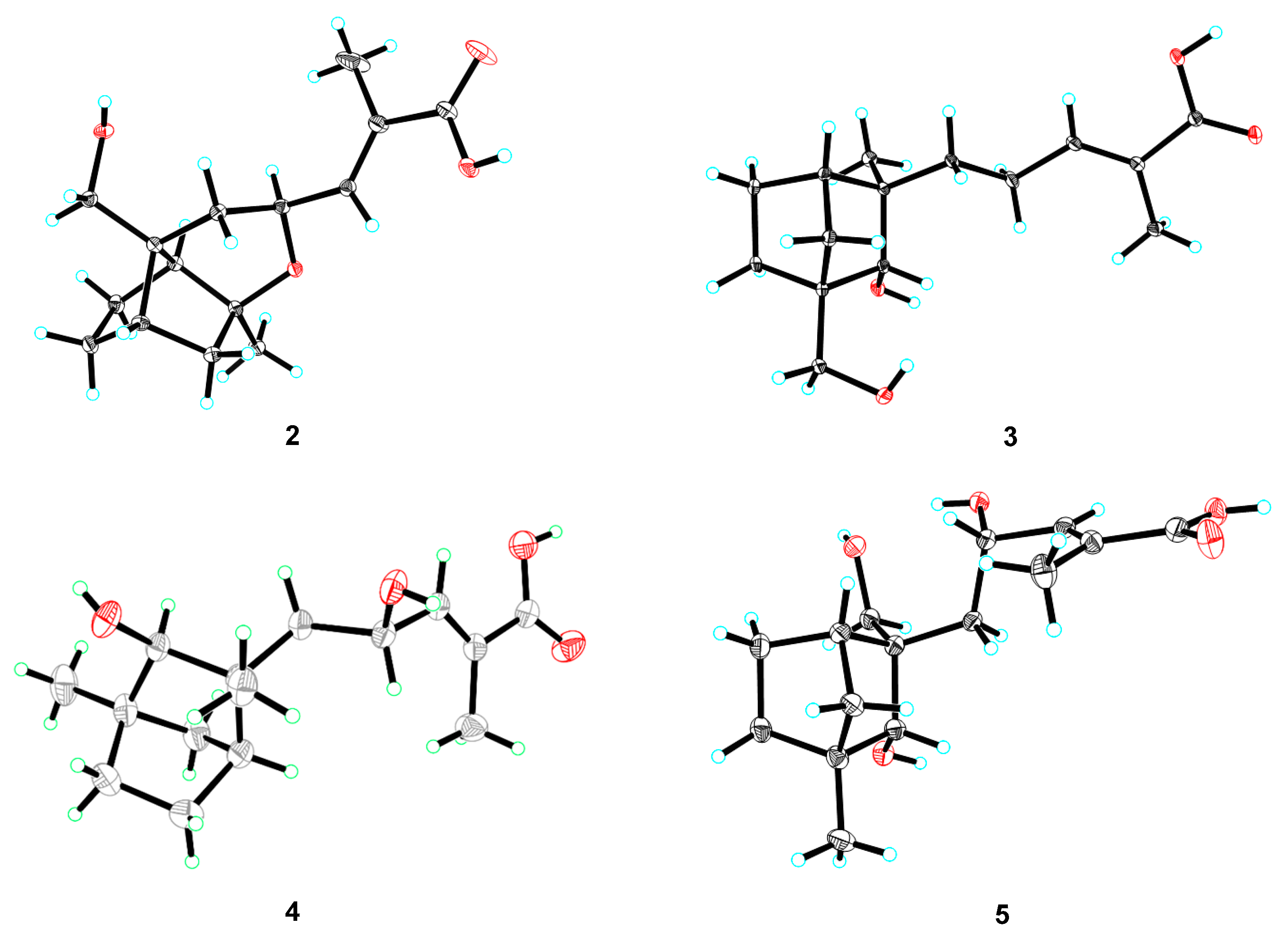
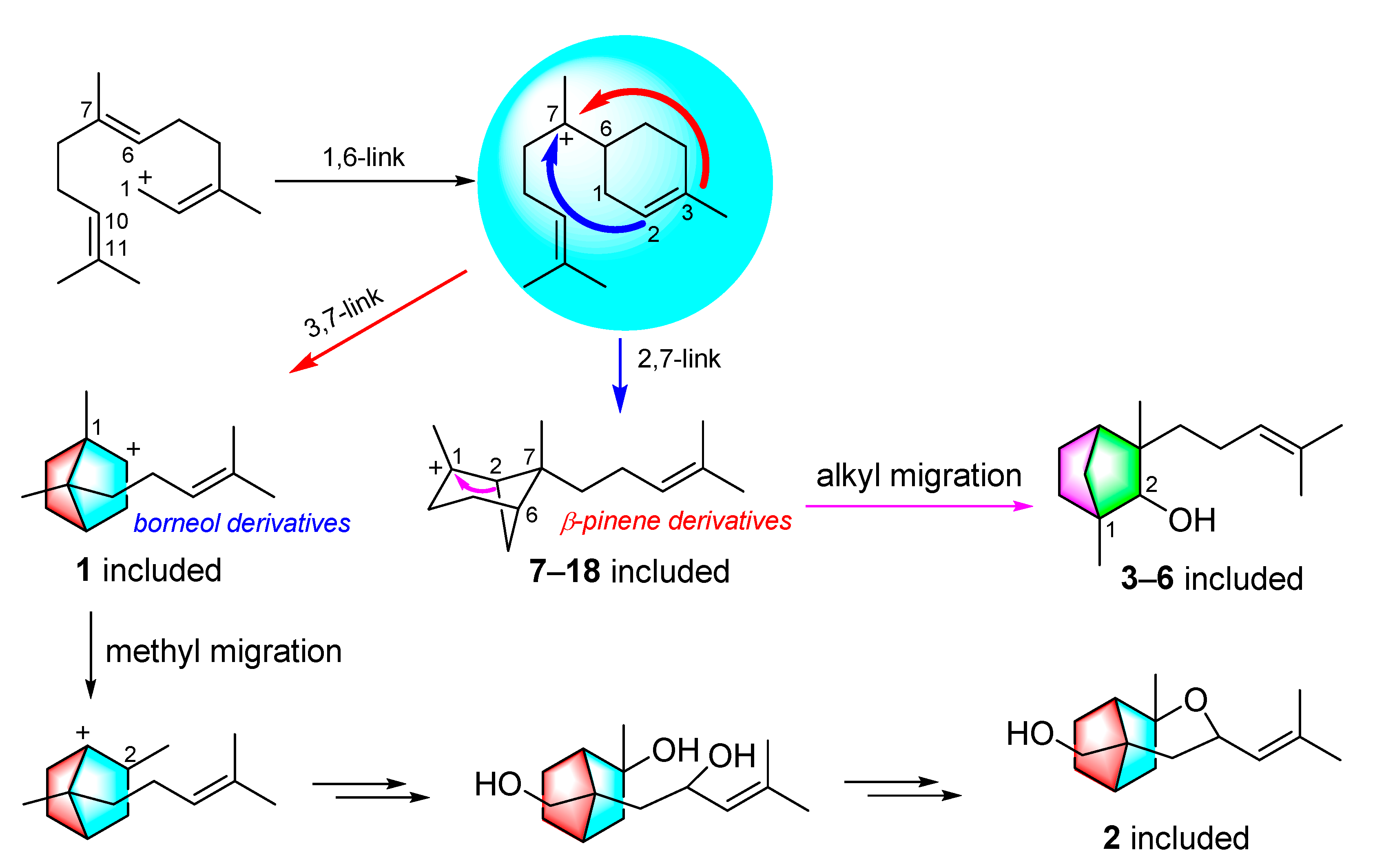
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Mo, M.Z.; Yang, L.; Mi, F.; Cao, Y.; Liu, C.L.; Tang, X.Z.; Wang, P.F.; Xu, J.P. Exploring the species diversity of edible mushrooms in Yunnan, Southwestern China, by DNA barcoding. J. Fungi 2021, 7, 310. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Cai, J.L.; Li, X.M.; Zhou, Z.Y.; Li, Z.H.; Liu, J.K. Chemical constituents and their bioactivities of mushroom Phellinus rhabarbarinus. J. Agric. Food Chem. 2016, 64, 1945–1949. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, S.B.; Chen, H.P.; Zhao, Z.Z.; Zhou, Z.Y.; Li, Z.H.; Feng, T.; Liu, J.K. New acetylenic acids and derivatives from the edible mushroom Craterellus lutescens (Cantharellaceae). J. Agric. Food Chem. 2017, 65, 3835–3841. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.P.; Zhao, Z.Z.; Li, Z.H.; Huang, Y.; Zhang, S.B.; Tang, Y.; Yao, J.N.; Chen, L.; Isaka, M.; Feng, T.; et al. Anti-proliferative and anti-inflammatory lanostane triterpenoids from the Polish edible mushroom Macrolepiota procera. J. Agric. Food Chem. 2018, 66, 3146–3154. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.R.; Li, T.H.; Jiang, Z.D.; Deng, W.Q.; Huang, H. A new yellow species of Craterellus (Cantharellales, Hydnaceae) from China. Phytotaxa 2018, 360, 35–44. [Google Scholar] [CrossRef]
- Bijeesh, C.; Manoj Kumar, A.; Vrinda, K.B.; Pradeep, C.K. Two new species of Craterellus (Cantharellaceae) from tropical India. Phytotaxa 2018, 346, 157–168. [Google Scholar] [CrossRef]
- Dahlman, M.; Danell, E.; Spatafora, J.W. Molecular systematics of Craterellus: Cladistic analysis of nuclear LSU rDNA sequence data. Mycol. Res. 2000, 104, 388–394. [Google Scholar] [CrossRef]
- Tripathy, S.S.; Rajoriya, A.; Gupta, N. Nutritional and biochemical analysis of Craterellus odoratus: Rare chanterelle basidiomycetes from similipal biosphere reserve of odisha. Ann. Appl. Bio-Sci. 2015, 2, A1–A6. [Google Scholar]
- Zhao, Z.Z.; Zhao, K.; Chen, H.P.; Bai, X.; Zhang, L.; Liu, J.K. Terpenoids from the mushroom-associated fungus Montagnula donacina. Phytochemistry 2018, 147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shen, Y.; Wang, F.; Leng, Y.; Liu, J.K. Rare merosesquiterpenoids from basidiomycete Craterellus odoratus and their inhibition of 11β-hydroxysteroid dehydrogenases. Phytochemistry 2010, 71, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yao, J.N.; Bai, X.; Li, Z.H.; Dong, Z.J.; Liu, J.K. Two new 4,6-dimethyl-3,4-dihydrochromen-2-one derivatives from Craterellus odoratus. J. Asian Nat. Prod. Res. 2017, 19, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Feng, T.; Li, Z.H.; Liu, J.K. Five new polyketides from the basidiomycete Craterellus odoratus. Nat. Prod. Bioprospect. 2012, 2, 170–173. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Feng, T.; Li, Z.H.; Liu, J.K. Four new compounds from the basidiomycete Craterellus odoratus. J. Asian Nat. Prod. Res. 2012, 14, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.M.; Fang, C.A.; Yao, F.Q.; Yu, Y.; Shen, Y.; Hou, Z.N.; Wang, Z.; Zhang, W.; Shan, W.G.; Zhan, Z.J. Bergamotane sesquiterpenes with alpha-glucosidase inhibitory activity from the plant pathogenic fungus Penicillium expansum. Chem. Biodivers. 2017, 14, e1600184. [Google Scholar] [CrossRef]
- Oh, H.; Gloer, J.B.; Shearer, C.A. Massarinolins A−C: New bioactive sesquiterpenoids from the aquatic fungus Massarina tunicata. J. Nat. Prod. 1999, 62, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Gloer, J.B.; Koster, B.; Malloch, D. Decipinin A and decipienolides A and B: New bioactive metabolites from the coprophilous fungus Podospora decipiens. J. Nat. Prod. 2002, 65, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Xia, G.Y.; Wang, L.Y.; Ge, G.B.; Zhang, H.W.; Zhang, J.F.; Wu, Y.Z.; Lin, S. Purpurolide A, 5/5/5 spirocyclic sesquiterpene lactone in nature from the endophytic fungus Penicillium purpurogenum. Org. Lett. 2018, 20, 7341–7344. [Google Scholar] [CrossRef] [PubMed]
- Pringle, R.B.; Beaun, A.C. Isolation of victoxinine from cultures of Helminthosporium victoriae. Phytopathology 1960, 50, 324–325. [Google Scholar]
- Dorn, F.; Arigoni, D. Structure of victoxinine. J. Chem. Soc. Chem. Commun. 1972, 1, 1342–1343. [Google Scholar] [CrossRef]
- Pringle, R.B. Comparative biochemistry of the phytopathogenic fungus Helminthosporium. XVI. the production of victoxinine by H. sativum and H. victoriae. Can. J. Biochem. 1976, 54, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Tan, X.M.; Yang, J.; Guo, L.P.; Ding, G. Naturally occurring seco-sativene sesquiterpenoid: Chemistry and biology. J. Agric. Food Chem. 2020, 68, 9827–9838. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Hygiene 1938, 27, 493–497. [Google Scholar]
- Ochi, T.; Shibata, H.; Higuti, T.; Kodama, K.H.; Kusumi, T.; Takaishi, Y. Anti-Helicobacter pylori compounds from Santalum album. J. Nat. Prod. 2005, 68, 819–824. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon−proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gao, H.; Chen, X.; Cai, X.; Yang, L.; Guo, L.; Yao, X.; Che, Y. Brasilamides A–D: Sesquiterpenoids from the plant endophytic fungus Paraconiothyrium brasiliense. Eur. J. Org. Chem. 2010, 17, 3302–3306. [Google Scholar] [CrossRef]
- Kono, Y.; Takeuchi, S.; Daly, J.M. Isolation and structure of a new victoxinine derivative produced by Helminthosporium victoriae. Agric. Biol. Chem. 1983, 47, 2701–2702. [Google Scholar] [CrossRef]
- Rodríguez López, M.; Bermejo, F.A. Total synthesis of (+)-massarinolin B and (+)-4-epi-massarinolin B, fungal metabolites from Massarina tunicata. Tetrahedron 2006, 62, 8095–8102. [Google Scholar] [CrossRef]
- Cane, D.E. Enzymic formation of sesquiterpenes. Chem. Rev. 1990, 90, 1089–1103. [Google Scholar] [CrossRef]
- Fraga, B.M. Natural sesquiterpenoids. Nat. Prod. Rep. 2013, 30, 1226–1264. [Google Scholar] [CrossRef]
- Massias, M.; Rebuffat, S.; Molho, L.; Chiaroni, A.; Riche, C.; Bodo, B. Expansolides A and B: Tetracyclic sesquiterpene lactones from Penicillium expansum. J. Am. Chem. Soc. 1990, 112, 8112–8115. [Google Scholar] [CrossRef]
- MacíAs, F.A.; Varela, R.M.; Simonet, A.M.; Cutler, H.G.; Cutler, S.J.; Hill, R.A. Absolute configuration of bioactive expansolides A and B from Aspergillus fumigatus Fresenius. Tetrahedron Lett. 2003, 44, 941–943. [Google Scholar] [CrossRef]
- Zhang, L.H.; Feng, B.M.; Chen, G.; Li, S.G.; Sun, Y.; Wu, H.H.; Bai, J.; Hua, H.M.; Wang, H.F.; Pei, Y.H. Sporulaminals A and B: A pair of unusual epimeric spiroaminal derivatives from a marine-derived fungus Paraconiothyrium sporulosum YK-03. RSC. Adv. 2016, 6, 42361–42366. [Google Scholar] [CrossRef]
- Davis, D.C.; Walker, K.L.; Hu, C.; Zare, R.N.; Waymouth, R.M.; Dai, M. Catalytic carbonylative spirolactonization of hydroxycyclopropanols. J. Am. Chem. Soc. 2016, 138, 10693–10699. [Google Scholar] [CrossRef] [Green Version]
- Dangroo, N.A.; Singh, J.; Dar, A.A.; Gupta, N.; Chinthakindi, P.K.; Kaul, A.; Khuroo, M.A.; Sangwan, P.L. Synthesis of α-santonin derived acetyl santonous acid triazole derivatives and their bioevaluation for T and B-cell proliferation. Eur. J. Med. Chem. 2016, 120, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Duan, K.T.; He, S.J.; Wu, B.; Zheng, Y.S.; Ai, H.L.; Li, Z.H.; He, J.; Zuo, J.P.; Liu, J.K. Ophiorrhines A and B, two immunosuppressive monoterpenoid indole alkaloids from Ophiorrhiza japonica. Org. Lett. 2018, 20, 7926–7928. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 49.9, C | − | 43.5, CH | 2.25, d (4.3) | 55.8, C | − | 41.5, C | − |
| 2 | 76.3, CH | 4.02, d (9.1) | 81.8, C | − | 79.7, CH | 3.51, s | 83.5, CH | 3.44, s |
| 3a 3b | 37.6, CH2 | 2.16, m 1.00, dd, (13.3, 3.3) | 48.2, CH2 | 1.98, dd, (14.3, 4.3) 1.28, d, (13.7) | 43.2, C | − | 48.5, C | − |
| 4 | 41.9, CH | 1.79, m | 42.3, CH | 1.86, t, (4.3) | 47.1, CH | 1.83, m | 46.5, CH | 1.80, s |
| 5a 5b | 27.5, CH2 | 1.69, m 1.27, m | 20.3, CH2 | 1.73, m 1.51, m | 26.5, CH2 | 1.70, m 1.39, m | 25.8, CH2 | 1.68, m 1.41, m |
| 6a 6b | 25.8, CH2 | 1.97, m 1.23, m | 26.5, CH2 | 1.72, m 1.16, m | 21.9, CH2 | 1.62, m 1.10, m | 25.0, CH2 | 1.69, m 0.99, m |
| 7a 7b | 50.9, C | − | 52.5, C | − | 37.2, CH2 | 1.54, d, (10.1) 1.11, m | 40.4, CH2 | 1.48, m 1.11, d, (10.3) |
| 8a 8b | 30.9, CH2 | 1.46, td, (12.8, 12.3, 5.1) 1.17, td, (12.8, 12.3, 5.1) | 33.7, CH2 | 1.98, dd, (14.3, 4.3) 1.74, m | 42.3, CH2 | 1.36, m | 48.9, CH2 | 1.71, m 1.45, m |
| 9a 9b | 24.5, CH2 | 2.25, m 2.07, m | 66.4, CH | 4.64, m | 24.7, CH2 | 2.15, m | 65.3, CH | 4.58, m |
| 10 | 141.1, CH | 6.69, t, (7.4) | 141.0, CH | 6.48, d, (7.8) | 144.1, CH | 6.72, t, (7.4) | 144.6, CH | 6.57, d, (9.28) |
| 11 | 128.5, C | − | 129.4, C | − | 128.8, C | − | 126.6, C | − |
| 12 | 171.3, C | − | 171.2, C | − | 172.0, C | − | 170.4, C | − |
| 13 | 11.2, CH3 | 1.80, s | 11.8, CH3 | 1.84, s | 12.3, CH3 | 1.78, s | 11.4, CH3 | 1.84, d, (1.5) |
| 14 | 12.3, CH3 | 0.85, s | 22.4, CH3 | 1.25, s | 17.0, CH3 | 0.86, s | 18.5, CH3 | 1.07, s |
| 15a 15b | 15.5, CH3 | 0.93, s | 63.4, CH2 | 3.64, d, (11.0) 3.47, d, (11.0) | 65.1, CH2 | 3.59, d, (11.0) 3.51, d, (11.0) | 16.1, CH3 | 0.91, s |
| Entry | 5 | 6 | 7 | 8 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 49.9, C | − | 49.8, C | − | 52.0, CH | 2.64, t, (5.3, 10.8) | 49.1, CH | 2.79, t, (5.4) |
| 2 | 86.0, CH | 3.34, d (1.3) | 82.2, CH | 3.47, d (1.3) | 150.0, C | − | 151.0, C | − |
| 3a 3b | 47.3, C | − | 47.8, C | − | 23.1, CH2 | 2.61, m 2.33, m | 24.3, CH2 | 2.65, m 2.40, m |
| 4a 4b | 44.0, CH | 2.06, d, (3.8) | 44.7, CH | 2.23, d, (3.5) | 23.5, CH2 | 2.11, m 1.82, m | 24.7, CH2 | 1.96, m |
| 5a 5b | 27.0, CH2 | 1.71, m 1.47, m | 27.6, CH2 | 1.65, m 1.39, m | 39.4, CH | 2.32, m | 40.6, CH | 2.27, m |
| 6a 6b | 26.7, CH2 | 1.72, m 0.99, m | 26.7, CH2 | 1.66, m 0.99, m | 26.5, CH2 | 2.20, m 1.47, d, (9.7) | 29.1, CH2 | 2.28, m 1.42, m |
| 7a 7b | 41.9, CH2 | 1.38, d, (10.6) 1.13, d, (10.6) | 41.5, CH2 | 1.47, d, (9.7) 1.07, d, (9.7) | 54.7, C | − | 56.5, C | − |
| 8a 8b | 47.0, CH2 | 1.70, m 1.53, dd, (2.6, 14.8) | 80.5, CH | 3.39, d, (6.0) | 40.4, CH2 | 2.50, dd, (12.4, 9.7) 2.02, dd, (12.4, 4.2) | 39.4, CH2 | 2.46, dd, (6.7, 11.9) 1.92, dd, (9.5, 11.9) |
| 9 | 66.6, CH | 4.60, m | 70.6, CH | 4.38, dd, (9.1, 6.0) | 73.7, CH | 4.91, m | 75.7, CH | 4.90, s |
| 10 | 142.6, CH | 6.49, d (8.2) | 138.4, CH | 6.53, d (8.6) | 143.0, CH | 6.74, dd, (1.5, 8.0) | 144.2, CH | 6.65, d, (8.2) |
| 11 | 131.5, C | − | 135.7, C | − | 128.1, C | − | 130.2, C | − |
| 12 | 174.9, C | − | 176.1, C | − | 169.8, C | − | 171.3, C | − |
| 13 | 13.1, CH3 | 1.80, s | 14.3, CH3 | 1.86, s | 11.4, CH3 | 1.85, d, (1.5) | 12.8, CH3 | 1.85, s |
| 14a 14b | 64.0, CH2 | 3.64, d, (11.7) 3.53, d, (11.7) | 19.9, CH3 | 1.04, s | 106.4, CH | 4.59, s | 106.8, CH | 4.57, s |
| 15a 15b | 19.4, CH3 | 1.03, s | 12.2, CH3 | 0.81, s | 107.0, CH2 | 4.74, q, (1.8) 4.68, dt, (3.0, 1.5) | 107.8, CH2 | 4.66, s 4.63, s |
| OCH3 | − | − | − | − | 53.3, CH3 | 3.29, s | 55.4, CH3 | 3.28, s |
| Entry | 9 | 10 | 11 | 12 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 51.0, CH | 2.10, t, (5.4) | 48.0, CH | 2.29, t, (5.6) | 51.3, CH | 2.09, t (5.7) | 47.4, CH | 2.70, t, (5.3) |
| 2 | 87.4, C | − | 88.2, C | − | 87.3, C | − | 89.2, C | − |
| 3a 3b | 32.0, CH2 | 1.88, m 1.61, m | 27.4, CH2 | 1.92, m 1.56, td, (10.2, 4.2) | 72.2, CH | 3.63, d, (6.8) | 29.5, CH2 | 2.06, m 1.83, m |
| 4a 4b | 22.4, CH2 | 1.88, m 1.76, m | 22.0, CH2 | 1.91, m 1.82, m | 32.8, CH2 | 2.25, m 1.85, m | 22.4, CH2 | 1.95, m 1.81, m |
| 5 | 39.2, CH | 2.24, q, (5.4) | 39.2, CH | 2.32, m | 37.8, CH | 2.27, m | 40.2, CH | 2.43, q, (5.3) |
| 6a 6b | 22.2, CH2 | 2.13, m 1.50, d, (10.1) | 22.3, CH2 | 2.14, m 1.51, d, (10.2) | 21.7, CH2 | 2.11, m 2.05, m | 22.1, CH2 | 2.28, m 1.75, d, (10.9) |
| 7 | 55.3, C | − | 53.7, C | − | 54.7, C | − | 53.6, C | − |
| 8a 8b | 32.4, CH2 | 1.91, m 1.72, m | 40.2, CH2 | 2.01, dd, (14.2, 4.2) 1.84, m | 40.1, CH2 | 2.00, dd, (14.2, 4.0) 1.81, dd, (14.2, 4.0) | 43.4, CH2 | 3.15, q, (3.2) |
| 9a 9b | 24.5, CH2 | 2.19, m 2.10, m | 65.7, CH | 4.45, td, (8.7, 3.8) | 65.8, CH | 4.42, td, (9.0, 4.0) | 199.4, C | − |
| 10 | 141.7, CH | 6.77, t, (7.5) | 143.6, CH | 6.67, d, (7.7) | 141.7, CH | 6.57, d, (9.0) | 130.6, CH | 7.11, s |
| 11 | 127.9, C | − | 127.3, C | − | 129.3, C | − | 142.4, C | − |
| 12 | 170.4, C | − | 170.5, C | − | 172.2, C | − | 169.9, C | − |
| 13 | 11.0, CH3 | 1.80, s | 11.4, CH3 | 1.83, s | 11.8, CH3 | 1.83, s | 13.4, CH3 | 2.15, s |
| 14a 14b | 70.2, CH2 | 3.82, d, (8.8) 3.47, d, (8.8) | 71.7, CH2 | 3.94, d, (9.4) 3.63, d, (9.4) | 71.5, CH2 | 3.85, d, (9.4) 3.61, d, (9.4) | 179.3, C | − |
| 15a 15b | 24.0, CH3 | 1.24, s | 65.9, CH2 | 3.49, d, (11.4) 3.45, d, (11.4) | 21.0, CH3 | 1.31, s | 23.6, CH3 | 1.51, s |
| Entry | 13 | 14 | 15 | 16 | ||||
|---|---|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 40.3, CH | 2.04, m | 73.5, C | − | 43.2, CH | 2.48, d, (6.7) | 47.6, CH | 1.87, m |
| 2 | 39.2, CH | 1.96, q, (5.1) | 40.3, CH | 1.38, m | 149.4, C | − | 150.2, C | − |
| 3a 3b | 37.3, CH2 | 1.84, m 1.76, m | 29.6, CH2 | 1.86, m 0.93, m | 24.7, CH2 | 2.64, m 2.32, m | 23.5, CH2 | 2.63, m 2.33, m |
| 4a 4b | 23.9, CH2 | 3.16, m | 26.7, CH2 | 1.79, m 1.08, m | 30.2, CH2 | 1.95, m 1.75, m | 22.6, CH2 | 1.95, m 1.88, m |
| 5 | 37.3, CH | 2.13, m | 47.3, CH | 1.33, m | 74.9, C | − | 37.6, CH | 2.18, q, (5.4) |
| 6a 6b | 18.0, CH2 | 1.63, m 1.27, m | 25.9, CH2 | 1.92, m 1.12, dd, (3.7, 12.5) | 34.6, CH2 | 2.33, m 1.79, d, (9.5) | 25.8, CH2 | 2.33, m 1.47, d, (10.1) |
| 7 | 42.1, C | − | 48.4, C | − | 48.6, C | − | 46.7, C | − |
| 8a 8b | 22.9, CH2 | 2.01, d, (5.7) 1.37, m | 38.1, CH2 | 1.53, t, (1.5) | 32.3, CH2 | 1.82, m 1.68, m | 29.9, CH2 | 1.87, m |
| 9a 9b | 23.7, CH | 1.84, m | 22.3, CH2 | 2.21, m | 23.8, CH2 | 2.28, m 2.21, m | 23.5, CH2 | 2.33, m 2.23, q, (8.0) |
| 10 | 140.7, CH | 6.71, dd, (7.4, 14.5) | 138.9, CH | 6.60, t, (6.6) | 142.5, CH | 6.81, t, (7.0) | 142.8, CH | 6.84, t, (8.0) |
| 11 | 129.3, C | − | 130.8, C | − | 127.8, C | − | 127.5, C | − |
| 12 | 171.3, C | − | 174.5, C | − | 171.3, C | − | 170.8, C | − |
| 13 | 11.4, CH3 | 1.81, s | 11.8, CH3 | 1.81, s | 11.1, CH3 | 1.82, s | 11.0, CH3 | 1.83, s |
| 14a 14b | 15.8, CH3 | 0.89, s | 22.0, CH3 | 1.09, s | 15.6, CH3 | 0.81, s | 61.1, CH2 | 3.43, d, (11.7) 3.33, d, (11.7) |
| 15a 15b | 65.3, CH2 | 3.34, dd, (2.6, 6.0) | 67.3, CH2 | 3.34, d, (3.3) | 106.1, CH2 | 4.63, d, (2.0) | 105.9, CH2 | 4.64, br s 4.60, br s |
| Entry | 17 | 18 | 19 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | δC, Type | δH (J in Hz) | |
| 1 | 48.2, CH | 3.00, t, (5.3, 10.6) | 45.0, CH | 2.03, t, (6.2) | 40.8, CH | 2.31, br s |
| 2 | 149.7, C | − | 44.2, CH | 1.94, q, (6.9) | 42.2, CH | 2.82, s |
| 3a 3b | 23.2, CH2 | 2.64, m 2.34, m | 96.5, C | − | 159.1, C | − |
| 4a 4b | 22.6, CH2 | 1.90, m | 41.0, CH2 | 2.24, m 1.87, d, (12.4) | 58.7, CH2 | 3.51, m 3.08, dd, (10.6, 2.1) |
| 5 | 38.5, CH | 2.21, m | 37.7, CH3 | 2.33, q, (6.2) | 58.8, CH2 | 3.51, m |
| 6a 6b | 44.1, C | − | 35.6, CH2 | 2.49, m 1.16, d, (9.7) | 58.7, CH | 1.99, br s |
| 7a 7b | 25.5, CH2 | 2.43, m 1.51, d, (10.0) | 39.2, C | − | 48.4, C | − |
| 8a 8b | 37.9, CH2 | 2.17, dd, (8.7, 15.0) 1.81, m | 39.5, CH2 | 1.85, m 1.71, dd, (14.8, 4.4) | 40.8, CH2 | 1.41, m 1.33, m |
| 9a 9b | 65.7, CH | 4.60, m | 65.7, CH | 4.45, m | 24.8, CH2 | 1.63, m 1.08, m |
| 10 | 143.1, CH | 6.65, d, (8.4) | 141.4, CH | 6.52, d, (8.2) | 44.7, CH | 1.08, m |
| 11 | 127.8, C | − | 129.7, C | − | 30.9, CH | 1.35, m |
| 12 | 171.6, C | − | 172.6, C | − | 20.2, CH3 | 0.87, d, (6.5) |
| 13 | 11.6, CH3 | 1.83, s | 12.0, CH3 | 1.85, s | 20.0, CH3 | 0.81, d, (6.5) |
| 14a 14b | 64.8, CH2 | 4.00, d, (11.9) 3.93, d, (11.9) | 69.3, CH2 | 4.01, d, (10.2) 3.94, d, (10.2) | 103.4, CH2 | 5.02, s 4.74, s |
| 15a 15b | 106.7, CH2 | 4.67, br s 4.64, br s | 13.8, CH3 | 1.06, d, (6.9) | 20.1, CH3 | 0.98, s |
| -OOCCH3 | 19.3, CH3 | 2.00, s | 45.0, CH | 2.03, t, (6.2) | 48.2, CH2 | 3.26, t, (6.0) |
| -OOCCH3 | 171.6, C | − | 44.2, CH | 1.94, q, (6.9) | 174.0, C | − |
| Entry | CC50 (μM) | Con A-Induced T-Cell Proliferation | LPS-Induced B-Cell Proliferation | ||
|---|---|---|---|---|---|
| IC50 (μM) | SI a | IC50 (μM) | SI a | ||
| 3 | >40 | − | − | 12.62 ± 1.14 | >3.17 |
| 10 | >40 | − | − | 19.40 ± 0.48 | >2.06 |
| 12 | >40 | − | − | 13.71 ± 0.65 | >2.92 |
| 13 | >40 | − | − | 15.43 ± 1.03 | >2.59 |
| 14 | >40 | − | − | 13.26 ± 1.29 | >3.02 |
| 15 | >40 | − | − | 17.12 ± 1.14 | >2.34 |
| 17 | >40 | 31.50 ± 1.79 | >1.27 | − | − |
| 19 | >40 | − | − | 22.68 ± 1.67 | >1.76 |
| 20 | >40 | 0.98 ± 0.01 | >40.82 | 0.67 ± 0.004 | >59.26 |
| 23 | >40 | − | − | 13.23 ± 0.97 | >3.02 |
| CsA | >2.80 | 0.04 | >70.00 | 0.47 | >5.95 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, Q.; Zhang, F.-L.; Li, Z.-H.; He, J.; Feng, T. Immunosuppressive Sesquiterpenoids from the Edible Mushroom Craterellus odoratus. J. Fungi 2021, 7, 1052. https://doi.org/10.3390/jof7121052
Dai Q, Zhang F-L, Li Z-H, He J, Feng T. Immunosuppressive Sesquiterpenoids from the Edible Mushroom Craterellus odoratus. Journal of Fungi. 2021; 7(12):1052. https://doi.org/10.3390/jof7121052
Chicago/Turabian StyleDai, Quan, Fa-Lei Zhang, Zheng-Hui Li, Juan He, and Tao Feng. 2021. "Immunosuppressive Sesquiterpenoids from the Edible Mushroom Craterellus odoratus" Journal of Fungi 7, no. 12: 1052. https://doi.org/10.3390/jof7121052