
Towards a Standardized Procedure for the Production of Infective Spores to Study the Pathogenesis of Dermatophytosis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Dermatophyte Strains
2.2. Growth Evaluation
2.3. Sporulation Evaluation
2.4. Production of Infective Spore Suspensions
2.5. Infection Development on Reconstructed Human Epidermis
2.5.1. Production of RHE and Infection by Dermatophytes
2.5.2. Histological Analysis
2.5.3. Measurement of Infection Level
2.5.4. Epidermal Barrier Integrity Monitoring
2.5.5. RNA Extraction, Reverse Transcription and Quantitative PCR
2.5.6. ELISA Assays
2.6. Statistical Analysis
3. Results
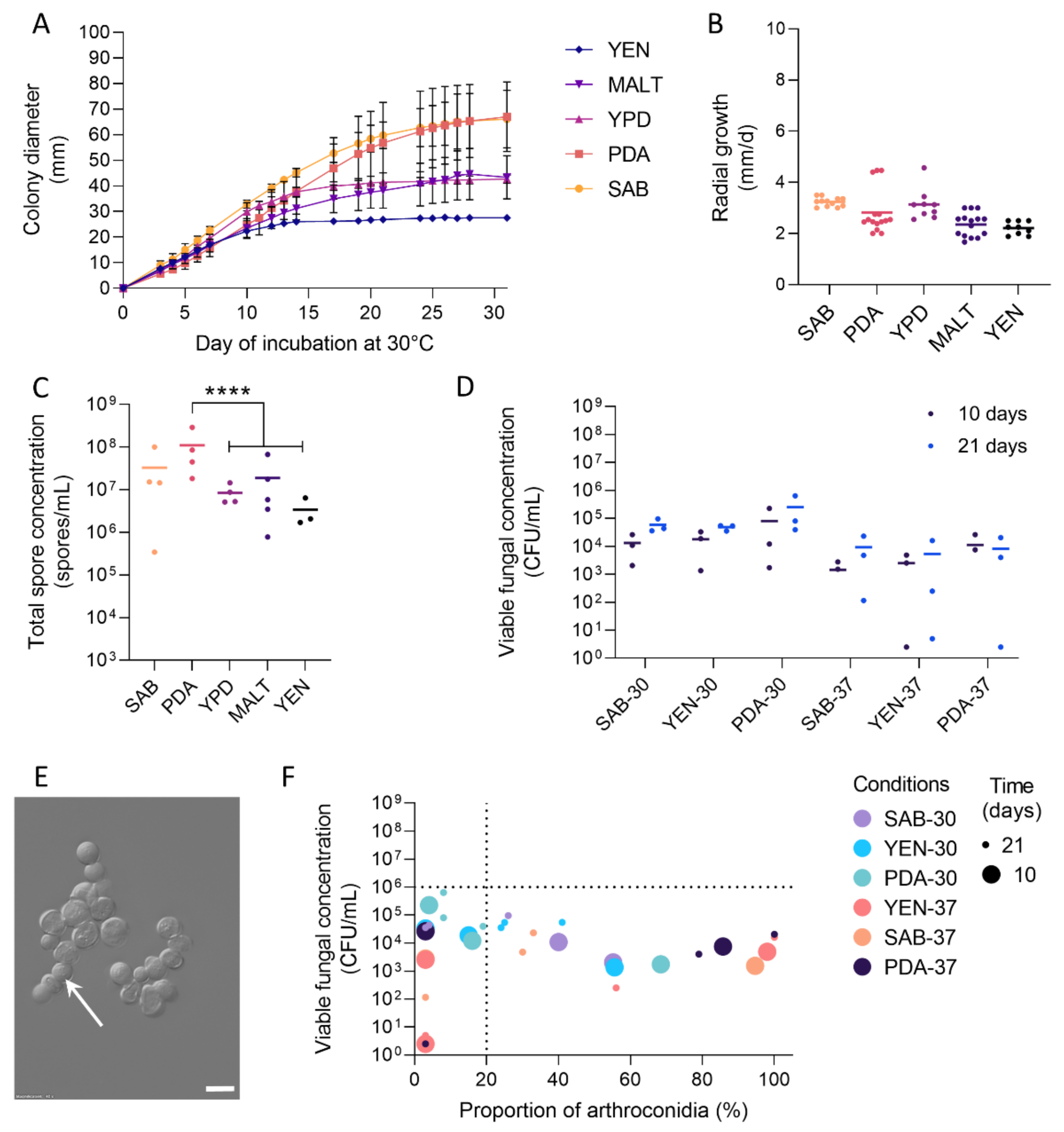
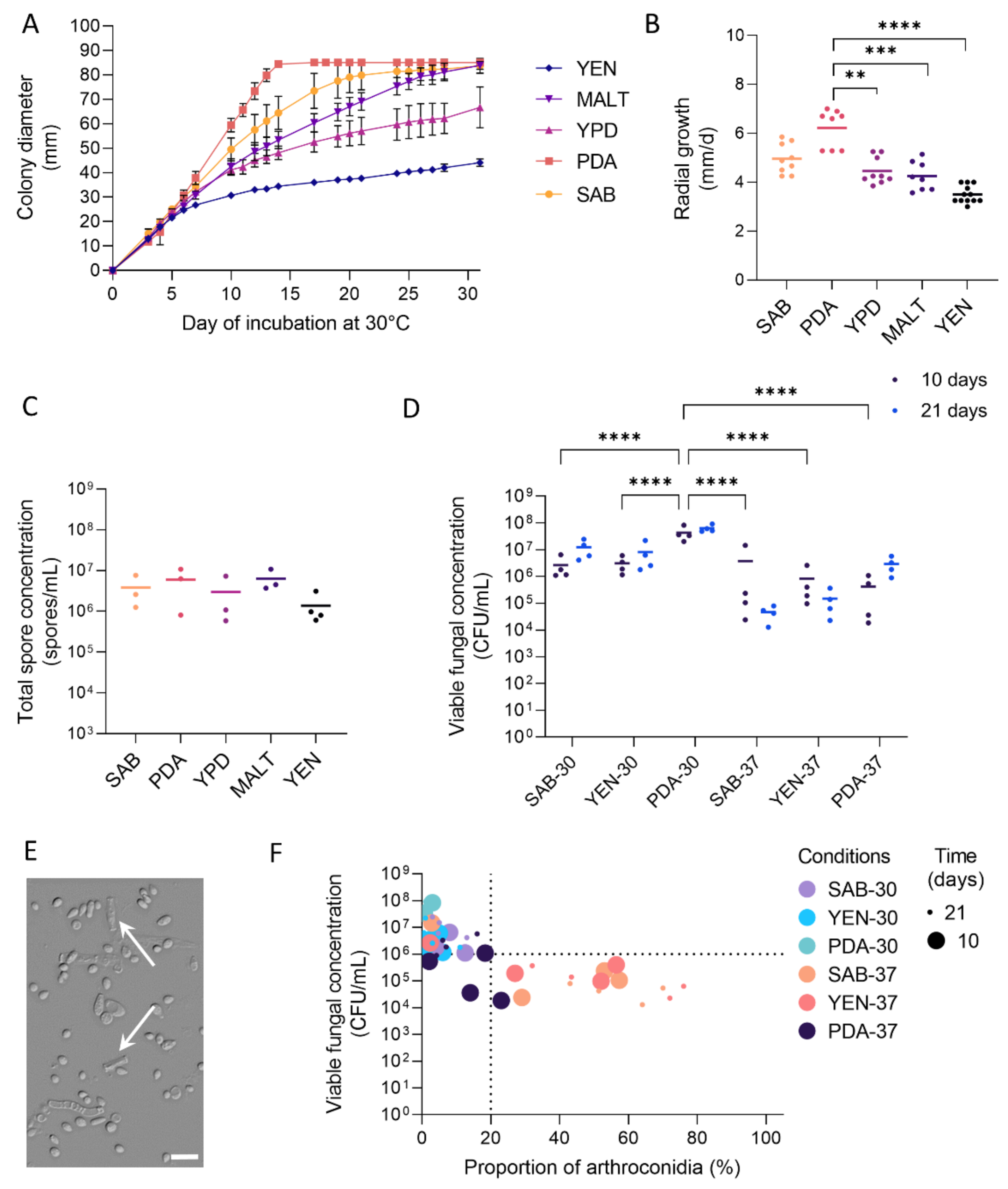
3.1. Growth and Sporulation of Dermatophytes Are Improved on PDA
3.2. High Viable Fungal Concentration Is Obtained after Incubation on PDA
3.3. Production of Arthroconidia Is Highly Variable According to Strains and Culture Conditions
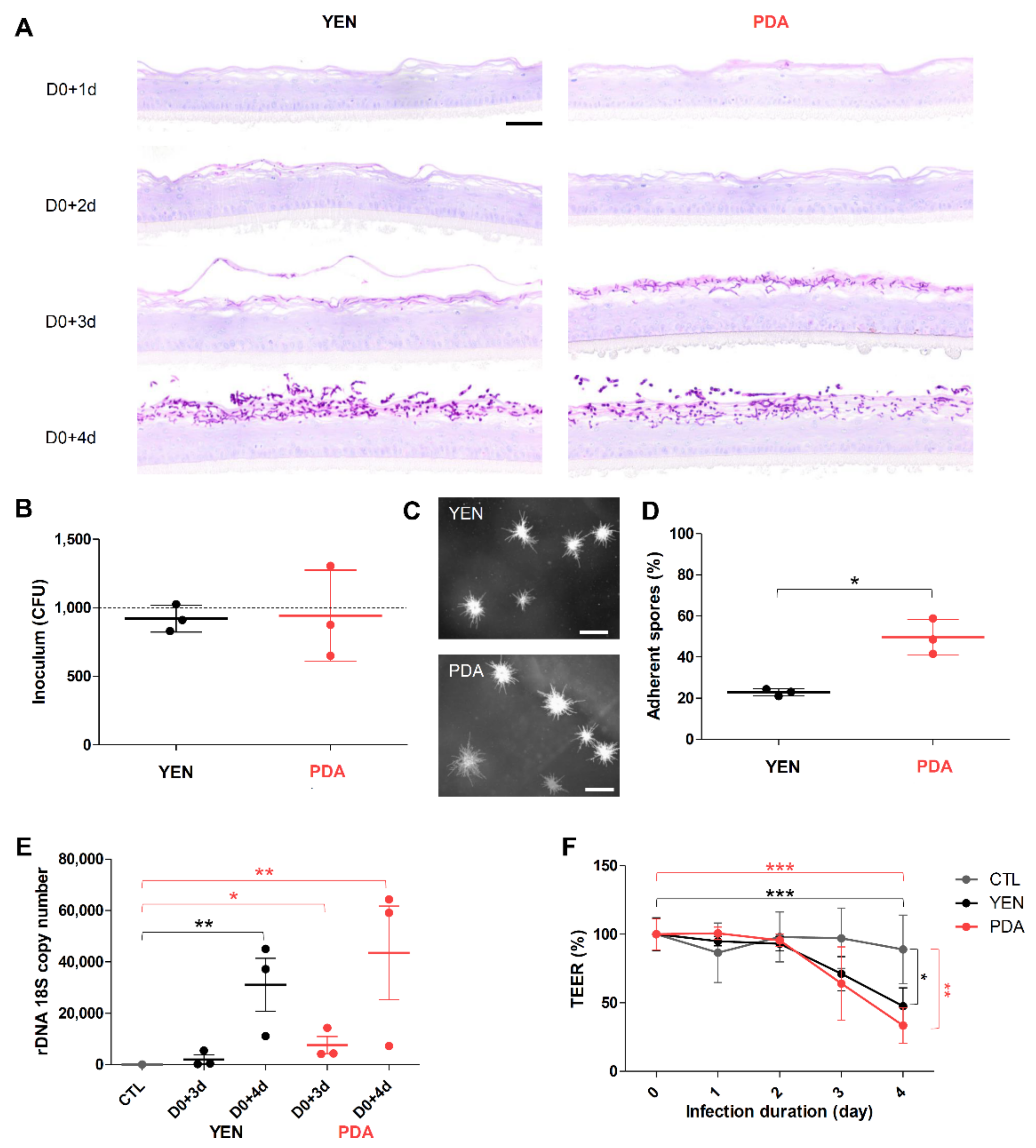
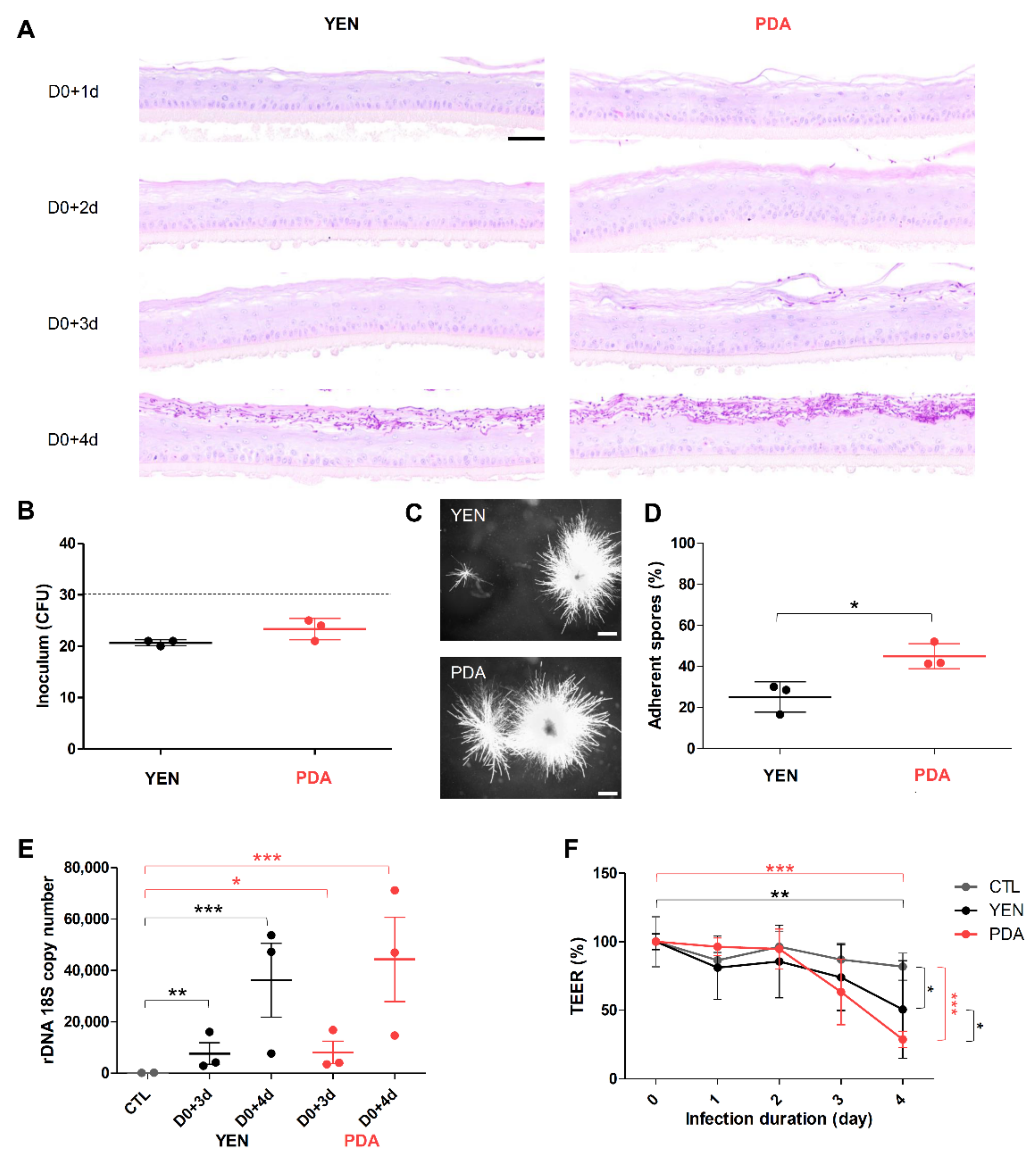
3.4. Spore Suspensions Recovered after 10 Days on PDA at 30 °C under 12% CO2 Are Adequate to Induce Infection on RHE
3.4.1. Adherence to RHE Is Improved for Spores Recovered on PDA
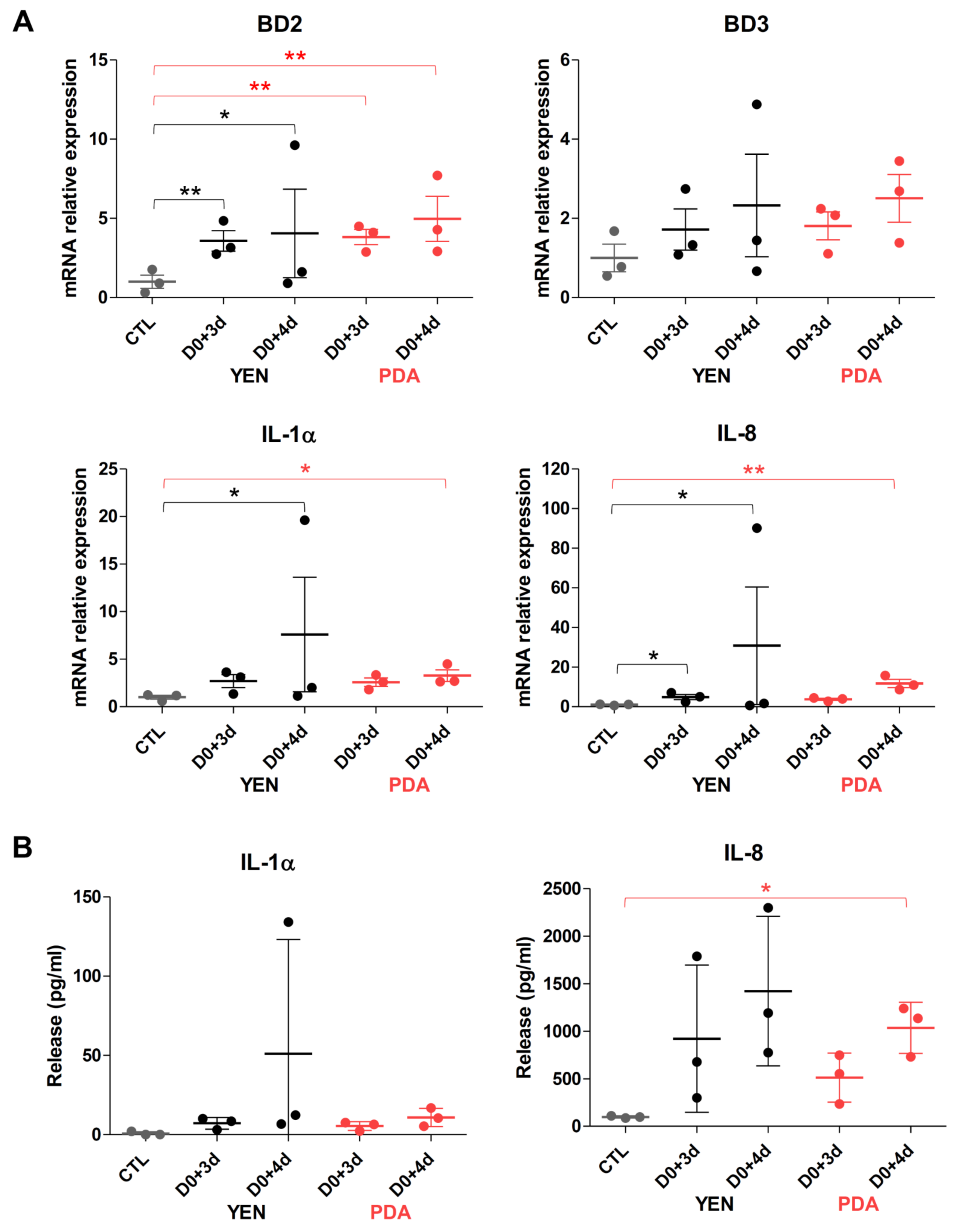
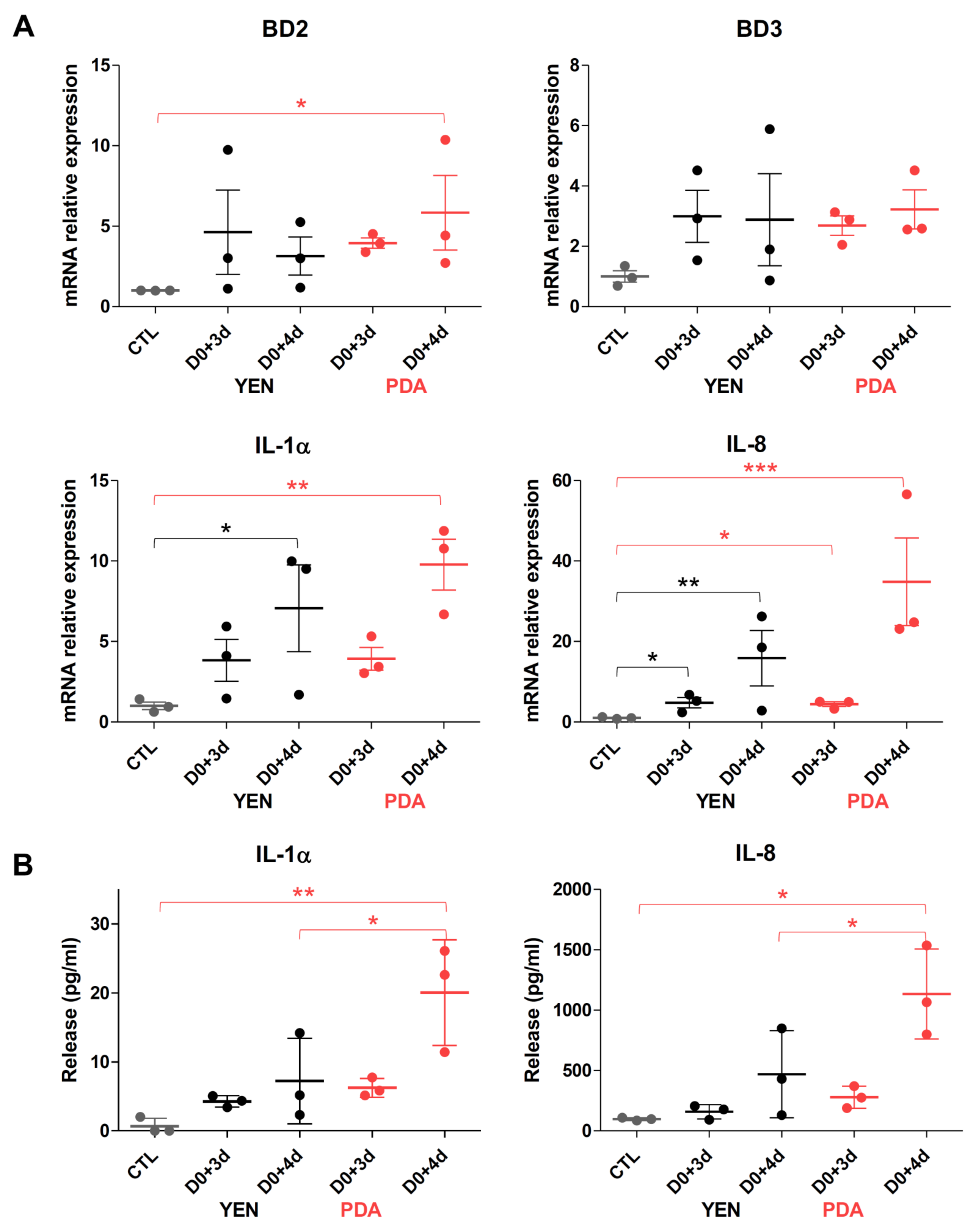
3.4.2. Responses of Keratinocytes to Infection Are More Consistent When Spores Are Produced on PDA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Weitzman, I.; Summerbell, R.C. The dermatophytes. Clin. Microbiol. Rev. 1995, 8, 240–259. [Google Scholar] [CrossRef]
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.M.; Holgersen, J.B.; Haedersdal, M.; Strauss, G.; Bitsch, M.; Svendsen, O.L.; Arendrup, M.C.; Svejgaard, E.L. Prevalence of toe nail onychomycosis in diabetic patients. Acta Derm. Venereol. 2006, 86, 425–428. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Daigle, D.; Foley, K.A. The prevalence of culture-confirmed toenail onychomycosis in at-risk patient populations. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Eckhard, M.; Lengler, A.; Liersch, J.; Bretzel, R.G.; Mayser, P. Fungal foot infections in patients with diabetes mellitus—Results of two independent investigations. Mycoses 2007, 50, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Sacheli, R.; Cuypers, L.; Seidel, L.; Darfouf, R.; Adjetey, C.; Lagrou, K.; Hayette, M.P. Epidemiology of dermatophytes in Belgium: A 5 years’ survey. Mycopathologia 2021, 186, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Gnat, S.; Lagowski, D.; Nowakiewicz, A. Genetic predisposition and its heredity in the context of increased prevalence of dermatophytoses. Mycopathologia 2021, 186, 163–176. [Google Scholar] [CrossRef]
- Gupta, A.K.; Foley, K.A.; Versteeg, S.G. New antifungal agents and new formulations against dermatophytes. Mycopathologia 2016, 182, 127–141. [Google Scholar] [CrossRef]
- Raschi, E.; Poluzzi, E.; Koci, A.; Caraceni, P.; Ponti, F.D. Assessing liver injury associated with antimycotics: Concise literature review and clues from data mining of the FAERS database. World J. Hepatol. 2014, 6, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monod, M.; Feuermann, M.; Salamin, K.; Fratti, M.; Makino, M.; Alshahni, M.M.; Makimura, K.; Yamada, T. Trichophyton rubrum azole resistance mediated by a new ABC transporter, TruMDR3. Antimicrob. Agents Chemother. 2019, 63, e00863-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagowski, D.; Gnat, S.; Nowakiewicz, A.; Osinska, M.; Dylag, M. Intrinsic resistance to terbinafine among human and animal isolates of Trichophyton mentagrophytes related to amino acid substitution in the squalene epoxidase. Infection 2020, 48, 889–897. [Google Scholar] [CrossRef]
- Kakurai, M.; Harada, K.; Maeda, T.; Hiruma, J.; Kano, R.; Demitsu, T. Case of tinea corporis due to terbinafine-resistant Trichophyton interdigitale. J. Dermatol. 2020, 47, e104–e105. [Google Scholar] [CrossRef]
- Gu, D.; Hatch, M.; Ghannoum, M.; Elewski, B.E. Treatment-resistant dermatophytosis: A representative case highlighting an emerging public health threat. JAAD Case Rep. 2020, 6, 1153–1155. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, V.; Bhattacharya, S.N.; Sharma, N.; Datt, S.; Kumar, P.; Rai, G.; Singh, P.K.; Taneja, B.; Das, S. Terbinafine resistance in dermatophytes: Time to revisit alternate antifungal therapy. J. Mycol. Med. 2021, 31, 101087. [Google Scholar] [CrossRef]
- Faway, E.; Lambert de Rouvroit, C.; Poumay, Y. In vitro models of dermatophyte infection to investigate epidermal barrier alterations. Exp. Dermatol. 2018, 27, 915–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambier, L.; Heinen, M.P.; Mignon, B. Relevant animal models in dermatophyte research. Mycopathologia 2017, 182, 229–240. [Google Scholar] [CrossRef]
- Achterman, R.R.; Moyes, D.L.; Thavaraj, S.; Smith, A.R.; Blair, K.M.; White, T.C.; Naglik, J.R. Dermatophytes activate skin keratinocytes via mitogen-activated protein kinase signaling and induce immune responses. Infect. Immun. 2015, 83, 1705–1714. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Huang, X.; Yi, J.; Chen, Z.; Ma, H.; Ye, C.; Chen, X.; Lai, W.; Chen, J. Trichophyton rubrum infection model based on the reconstructed human epidermis—Episkin. Chin. Med. J. 2016, 129, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Niu, Q.; Yu, X.; Jia, X.; Wang, J.; Lin, D.; Jin, Y. Assessment of the function of SUB6 in the pathogenic dermatophyte Trichophyton mentagrophytes. Med. Mycol. 2016, 54, 59–71. [Google Scholar]
- Burstein, V.L.; Guasconi, L.; Beccacece, I.; Theumer, M.G.; Mena, C.; Prinz, I.; Cervi, L.; Herrero, M.; Masih, D.T.; Chiapello, L.S. IL-17-mediated immunity controls skin infection and T Helper 1 response during experimental Microsporum canis dermatophytosis. J. Investig. Dermatol. 2018, 138, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Heinen, M.P.; Cambier, L.; Antoine, N.; Gabriel, A.; Gillet, L.; Bureau, F.; Mignon, B. Th1 and Th17 immune responses act complementarily to optimally control superficial dermatophytosis. J. Investig. Dermatol. 2019, 139, 626–637. [Google Scholar] [CrossRef] [Green Version]
- Baumbach, C.M.; Michler, J.K.; Nenoff, P.; Uhrlass, S.; Schrodl, W. Visualising virulence factors: Trichophyton benhamiae subtilisins demonstrated in a guinea pig skin ex vivo model. Mycoses 2020, 63, 970–978. [Google Scholar] [CrossRef]
- Baumbach, C.M.; Schrodl, W.; Nenoff, P.; Uhrlass, S.; Mulling, C.K.W.; Michler, J.K. Modeling dermatophytosis: Guinea pig skin explants represent a highly suitable model to study Trichophyton benhamiae infections. J. Dermatol. 2020, 47, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, S.; Matsuyama, T. Experimental tinea pedis induced by non-abrasive inoculation of Trichophyton mentagrophytes arthrospores on the plantar part of a guinea pig foot. J. Med. Vet. Mycol. 1987, 25, 203–213. [Google Scholar] [CrossRef]
- Tabart, J.; Baldo, A.; Vermout, S.; Nusgens, B.; Lapiere, C.; Losson, B.; Mignon, B. Reconstructed interfollicular feline epidermis as a model for Microsporum canis dermatophytosis. J. Med. Microbiol. 2007, 56, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Baldo, A.; Mathy, A.; Tabart, J.; Camponova, P.; Vermout, S.; Massart, L.; Marechal, F.; Galleni, M.; Mignon, B. Secreted subtilisin Sub3 from Microsporum canis is required for adherence to but not for invasion of the epidermis. Br. J. Dermatol. 2010, 162, 990–997. [Google Scholar] [CrossRef]
- Cambier, L.; Bagut, E.T.; Heinen, M.P.; Tabart, J.; Antoine, N.; Mignon, B. Assessment of immunogenicity and protective efficacy of Microsporum canis secreted components coupled to monophosphoryl lipid-A adjuvant in a vaccine study using guinea pigs. Vet. Microbiol. 2015, 175, 304–311. [Google Scholar] [CrossRef]
- Faway, E.; Cambier, L.; Mignon, B.; Poumay, Y.; Lambert de Rouvroit, C. Modeling dermatophytosis in reconstructed human epidermis: A new tool to study infection mechanisms and to test antifungal agents. Med. Mycol. 2017, 55, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faway, E.; Cambier, L.; De Vuyst, E.; Evrard, C.; Thiry, M.; Lambert de Rouvroit, C.; Mignon, B.; Poumay, Y. Responses of reconstructed human epidermis to Trichophyton rubrum infection and impairment of infection by the inhibitor PD169316. J. Investig. Dermatol. 2019, 139, 2080–2089. [Google Scholar] [CrossRef]
- Odds, F.; Ausma, J.; Van Gerven, F.; Woestenborghs, F.; Meerpoel, L.; Heeres, J.; Vanden Bossche, H.; Borgers, M. In vitro and in vivo activities of the novel antifungal agent r126638. Antimicrob. Agents Chemother. 2004, 48, 388–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staib, P.; Zaugg, C.; Mignon, B.; Weber, J.; Grumbt, M.; Pradervand, S.; Harshman, K.; Monod, M. Differential gene expression in the pathogenic dermatophyte Arthroderma benhamiae in vitro versus during infection. Microbiology 2010, 156, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Grumbt, M.; Defaweux, V.; Mignon, B.; Monod, M.; Burmester, A.; Wostemeyer, J.; Staib, P. Targeted gene deletion and in vivo analysis of putative virulence gene function in the pathogenic dermatophyte Arthroderma benhamiae. Eukaryot. Cell 2011, 10, 842–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambier, L.; Weatherspoon, A.; Defaweux, V.; Bagut, E.T.; Heinen, M.P.; Antoine, N.; Mignon, B. Assessment of the cutaneous immune response during Arthroderma benhamiae and A. vanbreuseghemii infection using an experimental mouse model. Br. J. Dermatol. 2014, 170, 625–633. [Google Scholar] [CrossRef]
- Singh, G.; Kumar, P.; Joshi, S.C. Treatment of dermatophytosis by a new antifungal agent ‘apigenin’. Mycoses 2014, 57, 497–506. [Google Scholar] [CrossRef]
- Tran, V.D.; De Coi, N.; Feuermann, M.; Schmid-Siegert, E.; Bagut, E.T.; Mignon, B.; Waridel, P.; Peter, C.; Pradervand, S.; Pagni, M.; et al. RNA sequencing-based genome reannotation of the dermatophyte Arthroderma benhamiae and characterization of its secretome and whole gene expression profile during infection. mSystems 2016, 1, 00036-16. [Google Scholar] [CrossRef] [Green Version]
- Rashid, A. Arthroconidia as vectors of dermatophytosis. Cutis 2001, 67, 23. [Google Scholar] [PubMed]
- Hashimoto, T.; Blumenthal, H.J. Factors affecting germination of Trichophyton mentagrophytes arthrospores. Infect. Immun. 1977, 18, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Hoff, B.; Schmitt, E.K.; Kuck, U. CPCR1, but not its interacting transcription factor AcFKH1, controls fungal arthrospore formation in Acremonium chrysogenum. Mol. Microbiol. 2005, 56, 1220–1233. [Google Scholar] [CrossRef]
- Bibel, D.J.; Crumrine, D.A.; Yee, K.; King, R.D. Development of arthrospores of Trichophyton mentagrophytes. Infect. Immun. 1977, 15, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, T.; Blumenthal, H.J. Survival and resistance of Trichophyton mentagrophytes arthrospores. Appl. Environ. Microbiol. 1978, 35, 274–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigl, E.; Hejtmanek, M. Differentiation of Trichophyton mentagrophytes arthrospores controlled by physical factors. Mycoses 1979, 22, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Wright, L.R.; Scott, E.M.; Gorman, S.P. Spore differentiation in a clinical strain of Trichophyton mentagrophytes. Microbios 1984, 39, 87–93. [Google Scholar]
- Yazdanparast, S.A.; Barton, R.C. Arthroconidia production in Trichophyton rubrum and a new ex vivo model of onychomycosis. J. Med. Microbiol. 2006, 55, 1577–1581. [Google Scholar] [CrossRef] [Green Version]
- Farnoodian, M.; Yazdanparast, S.A.; Sadri, M.F. Effects of environmental factors and selected antifungal agents on arthroconidia production in common species of Trichophyton genus and Epidermophyton floccosum. J. Biol. Sci. 2009, 9, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Alshahni, M.M.; Yamada, T.; Yo, A.; Murayama, S.Y.; Kuroda, M.; Hoshino, Y.; Ishikawa, J.; Watanabe, S.; Makimura, K. Insight into the draft whole-genome sequence of the dermatophyte Arthroderma vanbreuseghemii. Sci. Rep. 2018, 8, 15127. [Google Scholar] [CrossRef]
- De Vuyst, E.; Charlier, C.; Giltaire, S.; De Glas, V.; Lambert de Rouvroit, C.; Poumay, Y. Reconstruction of normal and pathological human epidermis on polycarbonate filter. Methods Mol. Biol. 2014, 1195, 191–201. [Google Scholar] [PubMed]
- Paugam, A.; L’Ollivier, C.; Viguie, C.; Anaya, L.; Mary, C.; de Ponfilly, G.; Ranque, S. Comparison of real-time PCR with conventional methods to detect dermatophytes in samples from patients with suspected dermatophytosis. J. Microbiol. Methods 2013, 95, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Minner, F.; Poumay, Y. Candidate housekeeping genes require evaluation before their selection for studies of human epidermal keratinocytes. J. Investig. Dermatol. 2009, 129, 770–773. [Google Scholar] [CrossRef] [Green Version]
- Faway, E.; Thiry, M.; Mignon, B.; Poumay, Y. Experimental models of dermatophytosis. In Dermatophytes and Dermatophytosis; Bouchara, J., Nenoff, P., Gupta, A.K., Chaturvedi, V., Eds.; Springer: Cham, Switzerland, 2021; pp. 135–160. [Google Scholar]
- Chin, B.; Knight, S.G. Growth of Trichophyton mentagrophytes and Trichophyton rubrum in increased carbon dioxide tensions. J. Gen. Microbiol. 1957, 16, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Emyanitoff, R.G.; Hashimoto, T. The effects of temperature, incubation atmosphere, and medium composition on arthrospore formation in the fungus Trichophyton mentagrophytes. Can. J. Microbiol. 1979, 25, 362–366. [Google Scholar] [CrossRef]
- Gupta, A.K.; Ahmad, I.; Porretta, M.; Summerbell, R.C. Arthroconidial formation in Trichophyton raubitschekii. Mycoses 2003, 46, 322–328. [Google Scholar] [CrossRef]
- Laurent, A.; Monod, M. Production of Trichophyton rubrum microspores in large quantities and its application to evaluate amorolfine/azole compound interactions in vitro. Mycoses 2017, 60, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Rippon, J.W.; Scherr, G.H. Induced dimorphism in dermatophytes. Mycologia 1959, 51, 902–914. [Google Scholar] [CrossRef]
- Zurita, J.; Hay, R.J. Adherence of dermatophyte microconidia and arthroconidia to human keratinocytes in vitro. J. Investig. Dermatol. 1987, 89, 529–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus—Species— Isolate Number | Isolation | |||
|---|---|---|---|---|
| Year | Lesion | Host | Country | |
| Microsporum | ||||
| M. audouinii IHEM 10316 | 1997 | Tinea capitis | Human | Belgium |
| M. canis IHEM 22957 | 2009 | Tinea | Cat | Belgium |
| M. canis IHEM 22958 | 2009 | Tinea | Cat | Belgium |
| M. canis IHEM 21239 | 2005 | Tinea | Cat | Belgium |
| Trichophyton | ||||
| T. benhamiae IHEM 20163 | 2002 | Tinea corporis | Human | Switzerland |
| T. benhamiae IHEM 20161 | 2002 | Tinea faciei | Human | Switzerland |
| T. interdigitale IHEM 00584 | 1981 | Tinea pedis | Human | Belgium |
| T. mentagrophytes IHEM 22740 | 2008 | Skin | Human | Switzerland |
| T. mentagrophytes IHEM 22733 | 2008 | Tinea | Cat | Switzerland |
| T. mentagrophytes IHEM 25841 | 2012 | Skin and hairs | Dog | Belgium |
| T. mentagrophytes TIMM 2789 1 | ||||
| T. rubrum IHEM 13894 | 1985 | Tinea cruris and onychomycosis | Human | Democratic Republic of the Congo |
| T. tonsurans IHEM 24958 | 2008 | Human | Belgium | |
| Composition in g/L | Sabouraud (SAB) | Potato Dextrose Agar (PDA) | Yeast Peptone Dextrose (YPD) | Malt Medium (MALT) | Yeast Extract Nitrogen (YEN) |
|---|---|---|---|---|---|
| Tryptone 1 | 10 | 20 | 6 | 10 | |
| Glucose 2 | 20 | 20 | |||
| Potato dextrose agar 3 | 39 | ||||
| Malt extract 4 | 20 | ||||
| Yeastextract 5 | 10 | 20 | |||
| Agar 6 | 20 | 5 | 20 | 20 | 20 |
| pH | 7.5 ± 0.3 | 5.4 ± 0.5 | 7.5 ± 0.1 | 6.1 ± 0.2 | 7.3 ± 0.2 |
| Gene Symbol | Forward Primer | Reverse Primer |
|---|---|---|
| BD2 | 5′-ATCAGCCATGAGGGTCTTGT-3′ | 5′-GAGACCACAGGTGCCAATTT-3′ |
| BD3 | 5′-TCCAGGTCATGGAGGAATCAT-3′ | 5′-CGAGCACTTGCCGATCTGT-3′ |
| IL-1α | 5′-AACCAGTGCTGCTGAAGGAGAT-3′ | 5′-TGGTCTCACTACCTGTGATGGTTT-3′ |
| IL-1β | 5′-TCCCCAGCCCTTTTGTTGA-3′ | 5′-TTAGAACCAAATGTGGCCGTG-3′ |
| IL-8 | 5′-GCAGAGGGTTGTGGAGAAGTTT-3′ | 5′-TTGGATACCACAGAGAATGAATTTTT-3′ |
| LL-37 | 5′-CCAGGACGACACAGCAGTCA-3′ | 5′-CTTCACCAGCCCGTCCTTC-3′ |
| RPLP0 | 5′-ATCAACGGGTACAAACGAGTC-3′ | 5′-CAGATGGATCAGCCAAGAAGG-3′ |
| TNFα | 5′-GAGGCCAAGCCCTGGTATG-3′ | 5′-CGGGCCGATTGATCTCAGC-3′ |
| Genus—Species— Isolate Number | Incubation Duration (Days) | Proportion of Arthroconidia (%) | |
|---|---|---|---|
| Min | Max | ||
| Microsporum | |||
| M. audouinii IHEM 10316 | 10 | nd 1 | nd |
| 21 | nd | nd | |
| 31 | nd | 67 | |
| M. canis IHEM 22957 | 21 | nd | 17 |
| M. canis IHEM 22958 | 21 | 3 | 67 |
| M. canis IHEM 21239 | 21 | 1 | 82 |
| Trichophyton | |||
| T. benhamiae IHEM 20161 | 10 | 1 | 21 |
| T. benhamiae IHEM 20163 | 10 | 1 | 1 |
| T. interdigitale IHEM 00584 | 10 | 1 | 9 |
| T. mentagrophytes IHEM 22733 | 10 | 14 | 24 |
| T. mentagrophytes IHEM 22740 | 10 | 30 | 66 |
| T. mentagrophytes IHEM 25841 | 10 | 1 | 15 |
| T. mentagrophytes TIMM 2789 | 10 | 3 | 9 |
| T. rubrum IHEM 13894 | 10 | 4 | 68 |
| T. tonsurans IHEM 24958 | 10 | 60 | 100 |
| 21 | 29 | 100 | |
| 31 | 72 | 93 | |
| Step | Procedure | Aim | Comment |
|---|---|---|---|
| Preculture | Incubation for 14 days on SAB at 30 °C | Growth of the starting fungal material | |
| Recovering of fungal material by scraping and suspension in PBS | |||
| Sporulation | Seeding on PDA and incubation for 10 days at 30 °C under 12% CO2 | Production of unicellular spores, mainly microconidia, and to a lesser extent arthroconidia | 21 days required for Microsporum spp. |
| Recovering of fungal material by scraping and suspension in PBS | |||
| Agitation | Four hours at 4 °C using magnetic stirrer | Separation of unicellular spores from hyphae | One-minute vortexing for fast procedure (reduced efficiency) |
| Filtration | Through three Miracloth layers | Recovery of unicellular elements only | Short hyphae can sometimes pass through the Miracloth filter |
| Washing | Centrifugation of the spore suspension at 3000× g for 5 min, discarding of the supernatant and resuspension of the pellet in PBS | Elimination of medium residues | Repeat until obtention of a clear supernatant, and at least three times |
| Final suspension | Resuspension of the pellet in an appropriate volume of PBS depending of the size of the pellet and of the final spore concentration wanted | Store spore suspensions at 4 °C and use within one month | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faway, E.; Staerck, C.; Danzelle, C.; Vroomen, S.; Courtain, C.; Mignon, B.; Poumay, Y. Towards a Standardized Procedure for the Production of Infective Spores to Study the Pathogenesis of Dermatophytosis. J. Fungi 2021, 7, 1029. https://doi.org/10.3390/jof7121029
Faway E, Staerck C, Danzelle C, Vroomen S, Courtain C, Mignon B, Poumay Y. Towards a Standardized Procedure for the Production of Infective Spores to Study the Pathogenesis of Dermatophytosis. Journal of Fungi. 2021; 7(12):1029. https://doi.org/10.3390/jof7121029
Chicago/Turabian StyleFaway, Emilie, Cindy Staerck, Célya Danzelle, Sophie Vroomen, Christel Courtain, Bernard Mignon, and Yves Poumay. 2021. "Towards a Standardized Procedure for the Production of Infective Spores to Study the Pathogenesis of Dermatophytosis" Journal of Fungi 7, no. 12: 1029. https://doi.org/10.3390/jof7121029