1. Introduction
Nucleotide substitutions are pivotal in promoting molecular evolution, and understanding their dynamics would contribute to genetic studies, such as on phylogenic reconstruction [
1], the estimation of nucleotide substitution bias [
2], and the relationship between base substitutions and some diseases [
3,
4]. Spontaneous mutations occur slowly and cause genome changes via single-base substitutions, insertions, deletions, and duplications [
5,
6], while mutagen-induced mutations occur explosively in a short period, resulting in similar mutation spectra to spontaneous mutations [
7,
8,
9,
10].
Previous studies have extensively investigated the mutational effects of ultraviolet radiation (UVR), the most pervasive mutagen, on DNA. Solar radiation is the predominant causal factor for squamous cell carcinoma [
11]. UVR mainly produces DNA photoproducts such as cyclobutane pyrimidine dimer (CPD) and pyrimidine (6-4)pyrimidone (64PP) [
12], and some other photoproducts in lower amounts, such as Dewar valence isomers and 8-Oxo-7,8-dihydroguanine [
13]. Base changes occur after these photoproducts are incorrectly repaired. UVR has specific mutation spectra, the so-called UV signature. According to wavelength, UVR is divided into ultraviolet A (UVA; wavelength: 320–400 nm), ultraviolet B (UVB; wavelength: 290–320 nm), and ultraviolet C (UVC; wavelength: 100–290 nm) [
14]. Although UVA, UVB, and UVC can produce UV signature mutations, such as C-to-T transition and CC-to-TT tandem transition, they show differentiated mutagenicity on DNA [
15,
16,
17,
18]. The mutation spectra of UVB are more similar to those of UVC than UVA [
18], probably because the energy absorption of UVA, UVB, and UVC in DNA is different in terms of wavelength. The yields of CPD and 64PP are highest at approximately 260 nm, which is in the action spectra of UVC and parallel with the absorption spectra of DNA [
12]. UVA radiation cannot be sufficiently absorbed compared with UVB [
4], thus contributing to the differential mutagenicity between UVA and UVB. It is widely accepted that UVB and UVC are the main producers of CPD and 64PP and that UVA mainly causes mutations by producing reactive oxygen species [
4,
19,
20,
21]. In addition to base substitutions, deletions are the sparsest and least studied mutations induced by UVR. In earlier studies, researchers have explored deletions caused by double-strand breaks using single linear DNA fragments with defined sequences [
22,
23]; therefore, deletion-related studies have been limited.
Different protocols have been used to detect UV-induced mutations. In early studies, irradiated DNA was often treated with polymerase-exonuclease, T4 endonuclease V, piperidine, or dimer-specific endonuclease to estimate photoproduct distributions [
24,
25,
26]. As PCR technology spread after the 1980s, more sequence-defined DNA fragments [
27,
28,
29,
30] were sequenced to detect UV-induced genetic changes. Whole-genome sequencing technology has promoted the discovery of new and specific mutations, such as genomic-level and cancer-specific mutations [
31,
32]. Detecting mutations at the genomic level would unravel new mechanisms underlying specific phenotypes induced by UV.
Limited by technology or cost, previous UV-induced mutational experiments were performed with defined DNA sequences or in specific models [
25,
26,
30], which might have resulted in narrow mutational spectra and could not present a systematic perspective. From this viewpoint, we chose the oidia of
Flammulina filiformis as the material for mutational experiments, expecting wide mutational spectra.
F. filiformis can spontaneously produce large amounts of oidia when cultivated on a PDA Petri dish, and the oidia have the same genotype as their mother monokaryons, with a genome of approximately 37 Mb in size. We mutated the oidia of the monokaryon
F. filiformis strain WT583 using UV light. After recovery, 100 randomly selected mutants and WT583 were sent for whole-genome sequencing. We precisely detected and located base changes in the WT583 genome and analyzed two typical UV signature mutations. We also characterized the distribution of point mutations and found DNA fragment deletions in the genomes of mutants. Besides mutational experiments on human skin cancer cells and
Escherichia coli [
31,
32], our research is among the few studies on UV-induced mutations and whole-genome sequencing.
4. Discussion
In this study, we investigated typical UVR-induced mutations based on whole-genome sequencing. Transitions (C:G→T:A and A:T→G:C) occurred at the highest frequency, with C:G→T:A transitions accounting for 60.40%. This result supports previous studies from a genomics perspective. UV-induced C:G→T:A transitions always accounted for the highest proportion of total substitutions in both eukaryotic and prokaryotic cells [
30,
46,
47,
48]. UVR mainly produces the photoproducts CPD and (6-4)PP in DNA [
49,
50]. Then, CPD and (6-4)PP are repaired by polymerases, resulting in C:G→T:A transitions [
29,
51], which explains why C:G→T:A transitions are the predominant component of UV-induced mutations. Previous studies on UV-induced mutations primarily focused on C:G→T:A transitions, while very few groups reported on A:T→G:C transitions, which ranked second among the six mutation types. Kamiva et al. found that A:T→G:C transitions mainly resulted from the photoproduct T(6-4)T with adjacent adenines in both single-strand DNA and double-strand DNA [
52], corresponding to our results. Moreover, mutations are easier at the T(6-4)T site than at the T = T dimer site [
52,
53]. Thus, it would be meaningful to study A:T→G:C transitions.
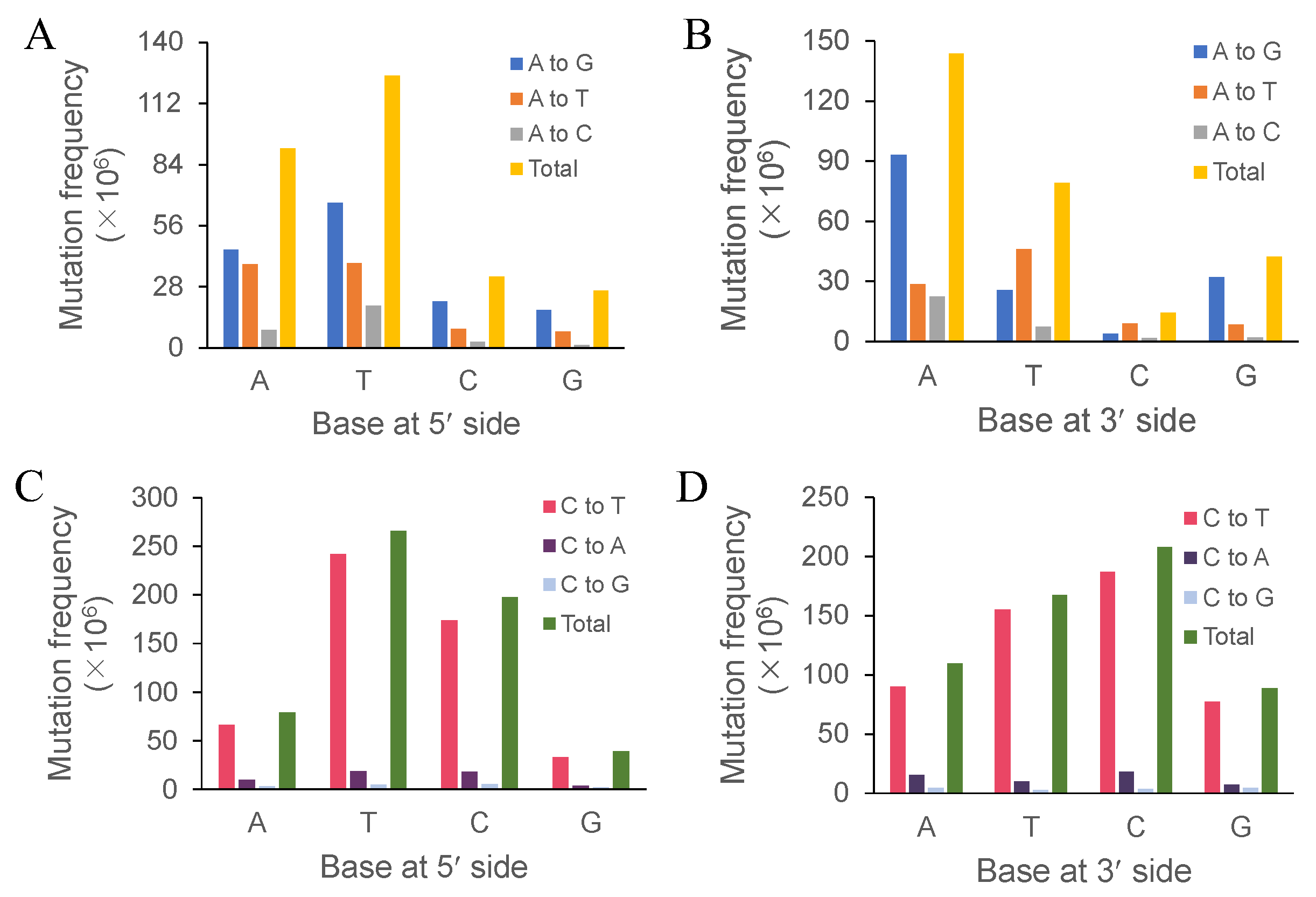
Both the immediately adjacent bases significantly influenced the base substitutions of the intermediate bases, while both the following bases showed much less influence [
54]. Thus, we focused on the effect of both the immediately adjacent bases. In a review of the previous literature, we found that researchers focused on the effects of bases at the 5′-side on the mutated bases [
55,
56], for they thought the effects of bases at the 3′-side were not significant. In our study, adenine at the 3′-side of adenine contributed more to adenine’s transition frequency than adenine at the 5′-side, which was also true for guanine at the 3′-side of adenine, and adenine, cytosine, and guanine at the 3′-side of cytosine. Therefore, we proposed that bases at the 3′-side would also significantly contribute to the transition frequency or the overall mutation frequency. A previous study obtained similar results to ours in bacterial genomes [
31]. Cytosine and thymine at the 3′-side might be conducive to the discrepancy we observed because cytosine and thymine belong to pyrimidine. This needs further evidence and discussion. Bases at the 3′-side noticeably affected the mutation frequency of the immediately adjacent bases. Future research should focus on the effect of bases at the 3′-side on base substitutions. The immediately adjacent bases of cytosine affected its transition—T > C > A > G and C > T > A > G, respectively—indicating that pyrimidines contributed more to the cytosine transition. In human skin cancer cells, UV-induced mutations featured C:G→T:A transitions at 5’-TCG-3’ [
4], which showed that the 5’-TC-3’ motif can result in a high cytosine transition frequency. Lee and Pfeifer’s study showed that the 5’-TCG-3’ and 5’-CCG-3’ motifs resulted in a high mutation frequency under UVR [
57]. Both studies showed that the 5’-TC-3’ motif increased the mutation frequency. In addition, our study showed that motifs 5’-TCT-3’ and 5’-TCC-3’ exhibited the highest mutation frequencies (352 and 348, respectively), while motifs 5’-TCG-3’ and 5’-CCG-3’ exhibited only 216 and 92 mutation sites (see
Supplementary Material S4), respectively. We ascribed the difference between their and our studies to the different DNA sequences, for they employed defined sequences or vectors with methylated or phosphorylated bases. Although different results were obtained by using altered DNA materials, it is widely accepted that base substitutions are significantly affected by the neighboring nucleotide composition [
58,
59]. The mutation-promoting influence of thymine on its nearest base may be because the A and T composition could decrease the repairing efficiency of DNA and stabilize the mismatch between guanine and adenine [
60,
61].
Limited by technology, earlier research often qualitatively visualized the distribution of UV-induced mutations on short and sequence-defined DNA fragments [
26,
27]. Next-generation sequencing technology and bioinformatics promote the visualization of mutation distributions in whole genomes. Hodis et al. applied whole-exome sequencing technology to quantitatively reveal mutation distributions in melanoma [
62]. Although they employed different methods, their results indicated non-random distributions in defined DNA fragments as well as whole genomes. Some factors affect the distribution of point mutations induced by UVR. Nucleosomes modulate the mutation distribution of both CPD and (6-4)PP. The photoproducts in DNA wrapped in nucleosomes showed a similar distribution pattern to naked DNA [
63]. Nucleosomes differentially influence the distributions of CPD and (6-4)PP. The distribution of (6-4)PP dimers is more random than that of CPDs or the total UV photoproducts, indicating that histones have different roles in the formation of CPD and (6-4)PP [
24]. The distribution of CPDs in the genome of cutaneous melanomas exhibits a periodical oscillation along with the periodical existence of histone octamers [
64]. The formation of CPD and (6-4)PP in specific base contexts also affects the distribution of mutations induced by UV light. In skin cancer cells, CPDs form at the PyrmCpG motif [
65], while (6-4)PP forms at 5′-TpC and 5′-CpC [
28]. These motifs are non-randomly distributed in DNA [
65]; therefore, these factors may contribute to the non-random distribution of mutations induced by UVR.
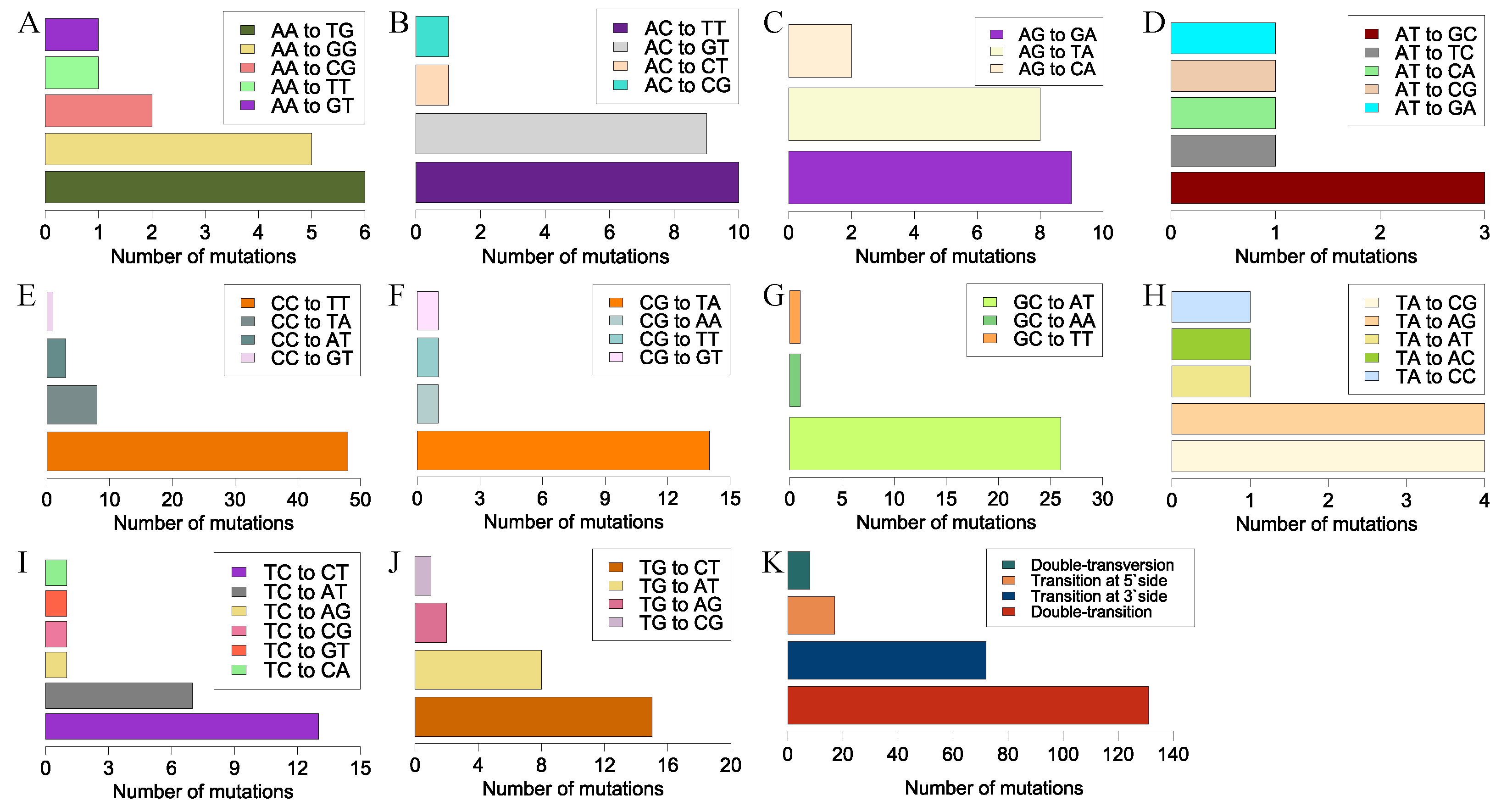
We observed another UV signature mutation, CC→TT, a typical tandem-base mutation. This well-known mutation in skin cells was generated by sunlight and frequently occurred at tandem pyrimidine sites [
66,
67,
68]. We also observed another tandem-base mutation, GC→AT, with a high mutation frequency, where both bases occurred as a transition. Previous studies showed that cytosine was prone to forming CPD with its 5′-side pyrimidine bases and, subsequently, CPD deaminated, resulting in a C→T transition with a mutation frequency reaching 80% [
4,
69]. Thus, CC→TT tandem mutations may result from double-deamination at CC sites. However, double-deamination may not explain why GC→AT occurred at a high frequency. Tandem mutations with 3′-side transitions occurred frequently; however, only the portion with a 5′-side T→A or A→T transversion held a higher mutation frequency. Compared with the portion with a low mutation frequency, a chemical bond number change at the 5′-side of the tandem base may affect the mutation frequency of such a tandem mutation. However, the underlying mechanism of this less-known tandem mutation type needs further investigation.
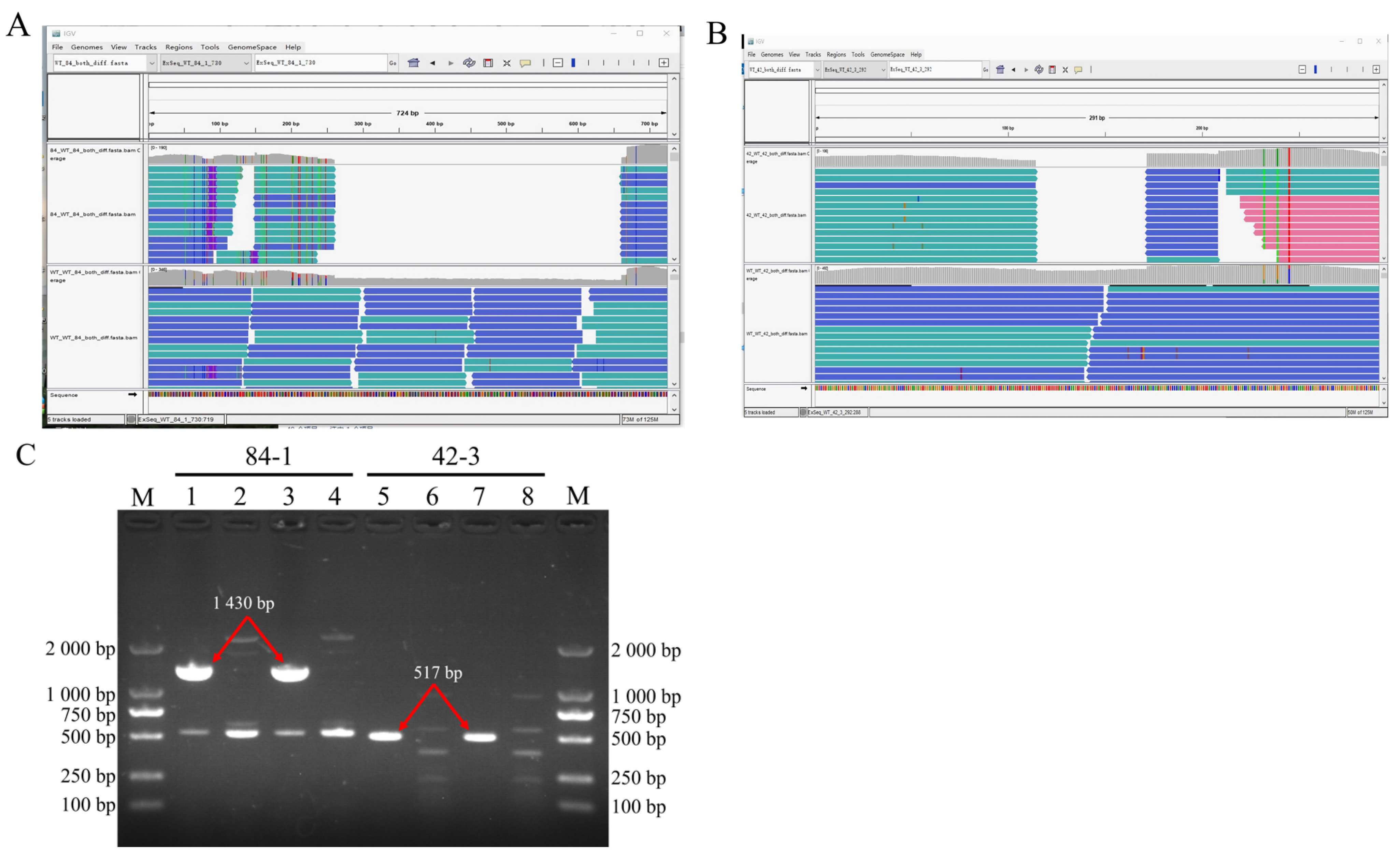
UV-induced DNA fragment deletions belong to structural variations (SVs), which include insertions, duplications, and copy number variations [
70]. SVs in the literature often exclusively refer to alterations of DNA fragments spanning from 50 bp to metabases [
21]. However, deletions under 200 bp were not detected and returned by the server in our studies, resulting in the incomplete detection of SV spectra. The deleted fragments caused by double-strand breaks usually have two different kinds of destinies, i.e., some are inserted back into the chromosome, while others drift away from the nucleus and are digested by nucleases [
71]. Our study showed that the deleted fragments were not inserted into the chromosome, as their corresponding k-mers were not visualized in the IGV window. Therefore, we proposed that both the deleted fragments might have been digested in the cytoplasm.
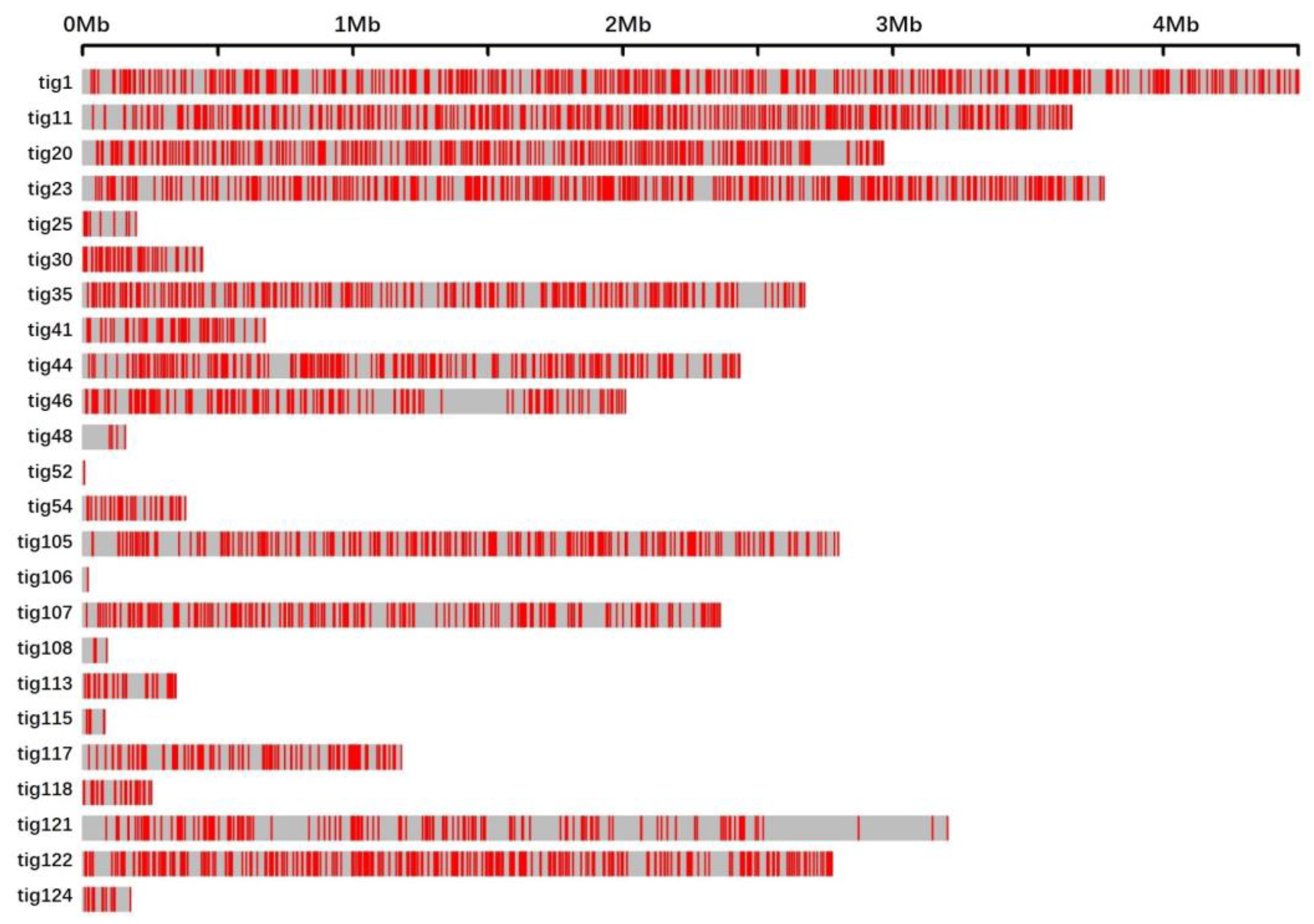
In conclusion, the genomes of 100 randomly selected mutants generated via UVR were sequenced. Subsequent analysis mainly focused on two kinds of UV signatures, i.e., single-base substitutions and tandem mutations. The immediately adjacent bases differentially affected the mutation frequencies of adenine and cytosine. Bases at the 3′-side also significantly influenced the transition frequency. Unlike the noticeable motif 5′-TCG-3′ in cancer induced by UV, motifs 5′-TCT-3′ and 5′-TCC-3′ in our studies exhibited the highest mutation frequencies. The sequence contexts of the tandem mutations affected the mutation frequency. The locations of all the 3707 single-base substitutions revealed a non-random distribution. Deletions, another rare type of UVR-induced mutation, were detected in only two mutants. The functional annotations of the non-synonymous genes indicated that UVR could be conducive to the mutational abundance in enoki mushrooms in a short period, which causes abundant phenotypic variations. Thus, UVR would be the best choice to produce mutants with visible phenotypic changes, or for other genetic studies, combined with its convenience and low cost.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}