

Hsp90-Mediated Multi-Drug Resistance in DNA Polymerase-Defective Strains of Candida albicans

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents, Strains, and Growth Conditions
2.2. Growth Curve Assay
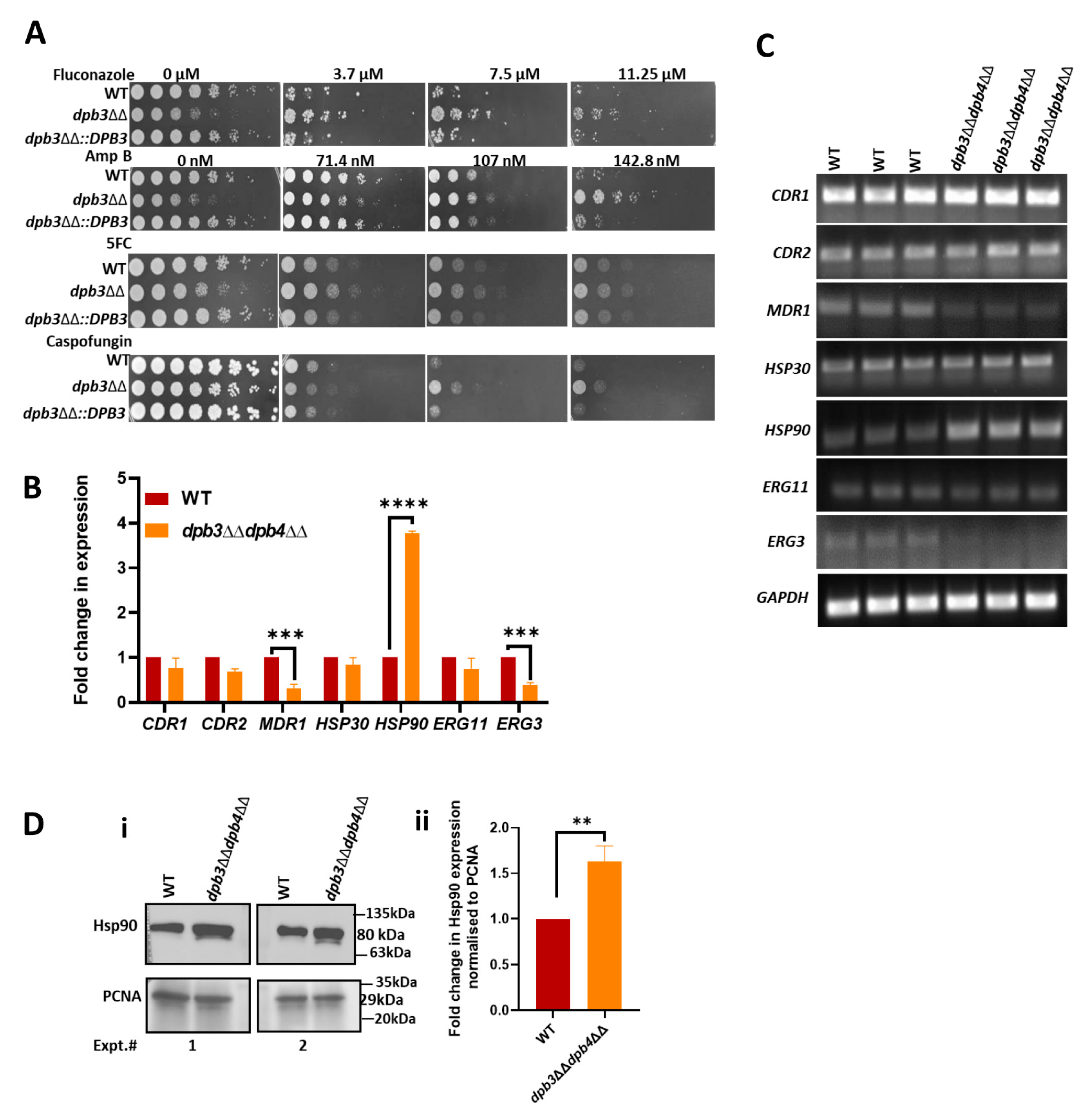
2.3. Antifungal Drug Susceptibility Assay by Spot Dilution Method
2.4. CFU Analysis
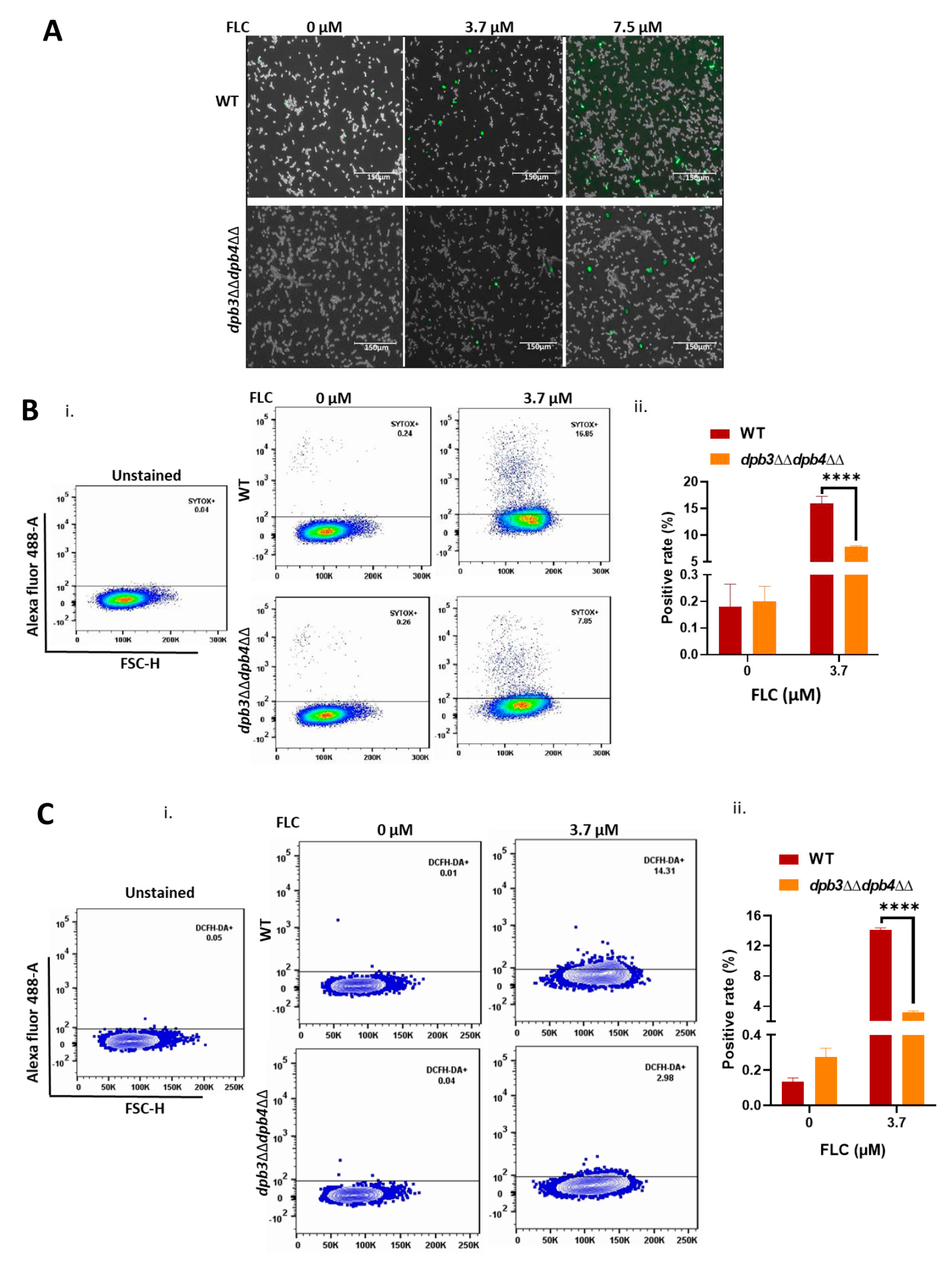
2.5. Membrane Permeability Assay
2.6. Measurement of ROS
2.7. Gene Expression Analysis by RT-PCR
2.8. Western Blot Analysis
2.9. Transmission Electron Microscopy
2.10. Cell Wall Components Estimation
2.11. Berberine Accumulation Assay
2.12. Biofilm Assay
2.13. Biofilm Detection by CLSM
2.14. Statistical Analysis
3. Results
3.1. Absence of Accessory Subunits of Polε Induces Azole Drug Resistance in C. albicans
3.2. Loss of Dpb3 and Dpb4 Subunits of Polε Also Induces Non-Azole Drug Resistance in C. albicans
3.3. Acquired-Mutagenesis-Independent Drug Resistance Mechanism in Polε-Defective Strain of C. albicans
3.4. Over-Expression of Hsp90 in Polε-Defective Strain of C. albicans
3.5. Multi-Drug Resistance Phenotype of dpb3/dpb4 Null Strains of C. albicans Is Due to Cell Wall Deformity but Independent of Tor1 Signaling
3.6. Hsp90 Function Is Critical for Multi-drug Resistance in C. albicans
3.7. Loss of Dpb3 and Dpb4 Subunits Induces Biofilm Formation of C. albicans
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef]
- Sahu, S.R.; Bose, S.; Singh, M.; Kumari, P.; Dutta, A.; Utkalaja, B.G.; Patel, S.K.; Acharya, N. Vaccines against candidiasis: Status, challenges and emerging opportunity. Front. Cell. Infect. Microbiol. 2022, 12, 1002406. [Google Scholar] [CrossRef]
- Peroumal, D.; Sahu, S.R.; Kumari, P.; Utkalaja, B.G.; Acharya, N. Commensal Fungus Candida albicans Maintains a Long-Term Mutualistic Relationship with the Host To Modulate Gut Microbiota and Metabolism. Microbiol. Spectr. 2022, 10, e0246222. [Google Scholar] [CrossRef]
- Desai, J.V.; Mitchell, A.P. Candida albicans Biofilm Development and Its Genetic Control. Microbiol. Spectr. 2015, 3, 1–12. [Google Scholar] [CrossRef]
- Rudkin, F.M.; Raziunaite, I.; Workman, H.; Essono, S.; Belmonte, R.; MacCallum, D.M.; Johnson, E.M.; Silva, L.M.; Palma, A.S.; Feizi, T.; et al. Single human B cell-derived monoclonal anti-Candida antibodies enhance phagocytosis and protect against disseminated candidiasis. Nat. Commun. 2018, 9, 5288. [Google Scholar] [CrossRef]
- Lee, Y.; Puumala, E.; Robbins, N.; Cowen, L.E. Antifungal Drug Resistance: Molecular Mechanisms in Candida albicans and Beyond. Chem. Rev. 2020, 121, 3390–3411. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.C.; Denning, D.W. The WHO fungal priority pathogens list as a game-changer. Nat. Rev. Microbiol. 2023, 21, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Banerjee, A.; Shah, A.H. Resistance to antifungal therapies. Essays Biochem. 2017, 61, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tu, J.; Han, G.; Liu, N.; Sheng, C. Heat shock protein 90 (Hsp90)/Histone deacetylase (HDAC) dual inhibitors for the treatment of azoles-resistant Candida albicans. Eur. J. Med. Chem. 2022, 227, 113961. [Google Scholar] [CrossRef] [PubMed]
- Iyer, K.R.; Robbins, N.; Cowen, L.E. The role of Candida albicans stress response pathways in antifungal tolerance and resistance. iScience 2022, 25, 103953. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.M.; Parker, J.E.; Bader, O.; Weig, M.; Gross, U.; Warrilow, A.G.S.; Rolley, N.; Kelly, D.E.; Kelly, S.L. Identification and characterization of four azole-resistant erg3 mutants of Candida albicans. Antimicrob. Agents Chemother. 2010, 54, 4527–4533. [Google Scholar] [CrossRef]
- Xiang, M.-J.; Liu, J.-Y.; Ni, P.-H.; Wang, S.; Shi, C.; Wei, B.; Ni, Y.-X.; Ge, H.-L. Erg11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res. 2013, 13, 386–393. [Google Scholar] [CrossRef]
- Flowers, S.A.; Barker, K.S.; Berkow, E.L.; Toner, G.; Chadwick, S.G.; Gygax, S.E.; Morschhäuser, J.; Rogers, P.D. Gain-of-function mutations in UPC2 Are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot. Cell 2012, 11, 1289–1299. [Google Scholar] [CrossRef]
- Tsao, S.; Rahkhoodaee, F.; Raymond, M. Relative Contributions of the Candida albicans ABC transporters Cdr1p and Cdr2p to clinical azole resistance. Antimicrob. Agents Chemother. 2009, 53, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- White, T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Coste, A.T.; Karababa, M.; Ischer, F.; Bille, J.; Sanglard, D. TAC1, Transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot. Cell 2004, 3, 1639–1652. [Google Scholar] [CrossRef]
- Schubert, S.; Rogers, P.D.; Morschhauser, J. Gain-of-function mutations in the transcription factor MRR1 are responsible for overexpression of the MDR1 efflux pump in fluconazole-resistant Candida dubliniensis strains. Antimicrob. Agents Chemother. 2008, 52, 4274–4280. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Sae-Tia, S.; Fries, B.C. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics 2020, 9, 312. [Google Scholar] [CrossRef]
- Perepnikhatka, V.; Fischer, F.J.; Niimi, M.; Baker, R.A.; Cannon, R.D.; Wang, Y.-K.; Sherman, F.; Rustchenko, E. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J. Bacteriol. 1999, 181, 4041–4049. [Google Scholar] [CrossRef]
- Czajka, K.M.; Venkataraman, K.; Brabant-Kirwan, D.; Santi, S.A.; Verschoor, C.; Appanna, V.D.; Singh, R.; Saunders, D.P.; Tharmalingam, S. Molecular Mechanisms Associated with Antifungal Resistance in Pathogenic Candida Species. Cells 2023, 12, 2655. [Google Scholar] [CrossRef]
- Vandeputte, P.; Ferrari, S.; Coste, A.T. Antifungal resistance and new strategies to control fungal infections. Int. J. Microbiol. 2012, 2012, 713687. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.A.; Gow, N.A.; Munro, C.A. Fungal echinocandin resistance. Fungal Genet. Biol. 2010, 47, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, P.; Chai, C.; Liu, J.; Sun, H.; Wu, Y.; Zhang, M.; Zhang, M.; Liu, X.; Yu, H. Structural and mechanistic insights into fungal β-1,3-glucan synthase FKS1. Nature 2023, 616, 190–198. [Google Scholar] [CrossRef]
- LaFayette, S.L.; Collins, C.; Zaas, A.K.; Schell, W.A.; Betancourt-Quiroz, M.; Gunatilaka, A.A.L.; Perfect, J.R.; Cowen, L.E. PKC Signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog. 2010, 6, e1001069. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Khandagale, P.; Thakur, S.; Sahu, J.K.; Utkalaja, B.G. Quaternary structural diversity in eukaryotic DNA polymerases: Monomeric to multimeric form. Curr. Genet. 2020, 66, 635–655. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Yeeles, J.T.P. An updated perspective on the polymerase division of labor during eukaryotic DNA replication. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 469–481. [Google Scholar] [CrossRef]
- Patel, S.K.; Sahu, S.R.; Utkalaja, B.G.; Bose, S.; Acharya, N. Pol32, an accessory subunit of DNA polymerase delta, plays an essential role in genome stability and pathogenesis of Candida albicans. Gut Microbes 2023, 15, 2163840. [Google Scholar] [CrossRef] [PubMed]
- Sahu, S.; Utkalaja, B.; Patel, S.; Acahrya, N. Spot Assay and Colony Forming Unit (CFU) Analyses–based sensitivity test for Candida albicans and Saccharomyces cerevisiae. Bio-Protocol 2023, 13, e4872. [Google Scholar] [CrossRef]
- Bose, S.; Singh, D.V.; Adhya, T.K.; Acharya, N. Escherichia coli, but Not Staphylococcus aureus, Functions as a Chelating Agent That Exhibits Antifungal Activity against the Pathogenic Yeast Candida albicans. J. Fungi 2023, 9, 286. [Google Scholar] [CrossRef]
- Manohar, K.; Peroumal, D.; Acharya, N. TLS dependent and independent functions of DNA polymerase eta (Polη/Rad30) from Pathogenic Yeast Candida albicans. Mol. Microbiol. 2018, 110, 707–727. [Google Scholar] [CrossRef]
- Acharya, N.; Prakash, L.; Prakash, S. Yeast 9-1-1 complex acts as a sliding clamp for DNA synthesis by DNA polymerase ε. J. Biol. Chem. 2023, 299, 102727. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Kondo, K.; Uehara, N.; Otokozawa, S.; Tsuji, N.; Yagihashi, A.; Watanabe, N. Endogenous reactive oxygen species is an important mediator of miconazole antifungal effect. Antimicrob. Agents Chemother. 2002, 46, 3113–3117. [Google Scholar] [CrossRef]
- Da Silva, A.R.; Batista de Andrade Neto, J.; da Silva, C.R.; de Sousa Campos, R.; Silva, R.A.C.; Freitas, D.D.; Aires do Nascimento, F.B.S.; Dantas de Andrade, L.N.; Sampaio, L.S.; Grangeiro, T.B.; et al. Berberine Antifungal Activity in Fluconazole-Resistant Pathogenic Yeasts: Action Mechanism Evaluated by Flow Cytometry and Biofilm Growth Inhibition in Candida spp. Antimicrob. Agents Chemother. 2016, 60, 3551–3557. [Google Scholar] [CrossRef]
- Li, Y.; Sun, S.; Guo, Q.; Ma, L.; Shi, C.; Su, L.; Li, H. In vitro interaction between azoles and cyclosporin A against clinical isolates of Candida albicans determined by the chequerboard method and time-kill curves. J. Antimicrob. Chemother. 2008, 61, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.P.; Nelson, Z.W.; Hetrick, E.D.E.; Gottschling, D. A genetic screen for increased loss of heterozygosity in Saccharomyces cerevisiae. Genetics 2008, 179, 1179–1195. [Google Scholar] [CrossRef]
- Araki, H.; Hamatake, R.K.; Morrison, A.; Johnson, A.L.; Johnston, L.H.; Sugino, A. Cloning DPB3, the gene encoding the third subunit of DNA polymerase II of Saccharomyces cerevisiae. Nucleic Acids Res. 1991, 19, 4867–4872. [Google Scholar] [CrossRef]
- Sundaram, R.; Manohar, K.; Patel, S.K.; Acharya, N.; Vasudevan, D. Structural analyses of PCNA from the fungal pathogen Candida albicans identify three regions with species-specific conformations. FEBS Lett. 2021, 595, 1328–1349. [Google Scholar] [CrossRef]
- Chun, J.; Ko, Y.-H.; So, K.-K.; Cho, S.-H.; Kim, D.-H. A fungal GPI-anchored protein gene functions as a virulence and antiviral factor. Cell Rep. 2022, 41, 111481. [Google Scholar] [CrossRef]
- Kapteyn, J.C.; Hoyer, L.L.; Hecht, J.E.; Muller, W.H.; Andel, A.; Verkleij, A.J.; Makarow, M.; Van Den Ende, H.; Klis, F.M. The cell wall architecture of Candida albicans wild-type cells and cell wall-defective mutants. Mol. Microbiol. 2000, 35, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, R.S.; Uppuluri, P.; Zaas, A.K.; Collins, C.; Senn, H.; Perfect, J.R.; Heitman, J.; Cowen, L.E. Hsp90 Orchestrates Temperature-Dependent Candida albicans Morphogenesis via Ras1-PKA Signaling. Curr. Biol. 2009, 19, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Cowen, L.E.; Singh, S.D.; Köhler, J.R.; Collins, C.; Zaas, A.K.; Schell, W.A.; Aziz, H.; Mylonakis, E.; Perfect, J.R.; Whitesell, L.; et al. Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Natl. Acad. Sci. USA 2009, 106, 2818–2823. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chang, W.; Zhang, M.; Li, X.; Jiao, Y.; Lou, H. Synergistic and drug-resistant reversing effects of diorcinol D combined with fluconazole against Candida albicans. FEMS Yeast Res. 2015, 15, fov001. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; An, M.; Shen, H.; Huang, X.; Yao, X.; Liu, J.; Zhu, F.; Zhang, S.; Chen, S.; He, L.; et al. The non-Geldanamycin Hsp90 inhibitors enhanced the antifungal activity of fluconazole. Am. J. Transl. Res. 2015, 7, 2589–2602. [Google Scholar] [PubMed]
- Robbins, N.; Uppuluri, P.; Nett, J.; Rajendran, R.; Ramage, G.; Lopez-Ribot, J.L.; Andes, D.; Cowen, L.E. Hsp90 governs dispersion and drug resistance of fungal biofilms. PLoS Pathog. 2011, 7, e1002257. [Google Scholar] [CrossRef] [PubMed]
- Finkel, J.S.; Mitchell, A.P. Genetic control of Candida albicans biofilm development. Nat. Rev. Genet. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Gulati, M.; Nobile, C.J. Candida albicans biofilms: Development, regulation, and molecular mechanisms. Microbes Infect. 2016, 18, 310–321. [Google Scholar] [CrossRef]
- Pistoia, E.S.; Cosio, T.; Campione, E.; Pica, F.; Volpe, A.; Marino, D.; Di Francesco, P.; Monari, C.; Fontana, C.; Favaro, M.; et al. All-Trans Retinoic Acid Effect on Candida albicans Growth and Biofilm Formation. J. Fungi 2022, 8, 1049. [Google Scholar] [CrossRef]
- Jensen, R.H.; Astvad, K.M.T.; Silva, L.V.; Sanglard, D.; Jørgensen, R.; Nielsen, K.F.; Mathiasen, E.G.; Doroudian, G.; Perlin, D.S.; Arendrup, M.C. Stepwise emergence of azole, echinocandin and amphotericin B multidrug resistance in vivo in Candida albicans orchestrated by multiple genetic alterations. J. Antimicrob. Chemother. 2015, 70, 2551–2555. [Google Scholar] [CrossRef]
- Acharya, N.; Klassen, R.; Johnson, R.E.; Prakash, L.; Prakash, S. PCNA binding domains in all three subunits of yeast DNA polymerase delta modulate its function in DNA replication. Proc. Natl. Acad. Sci. USA 2011, 108, 17927–17932. [Google Scholar] [CrossRef]
- Cowen, L.E.; Anderson, J.B.; Kohn, L.M. Evolution of Drug Resistance in Candida albicans. Annu. Rev. Microbiol. 2002, 56, 139–165. [Google Scholar] [CrossRef]
- Belenky, P.; Camacho, D.; Collins, J.J. Fungicidal drugs induce a common oxidative-damage cellular death pathway. Cell Rep. 2013, 3, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Casalongue, M.E.; Lee, J.; Diamond, A.; Shuldiner, S.; Moir, R.D.; Willis, I.M. Differential phosphorylation of a regulatory subunit of protein kinase CK2 by target of rapamycin complex 1 signaling and the Cdc-like kinase Kns1. J. Biol. Chem. 2015, 290, 7221–7233. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- McClellan, A.J.; Xia, Y.; Deutschbauer, A.M.; Davis, R.W.; Gerstein, M.; Frydman, J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 2007, 131, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, Z.; Shi, X.; Liu, L.; Zhang, P.; Golemis, E.A.; Tu, Z. HSP90 inhibition downregulates DNA replication and repair genes via E2F1 repression. J. Biol. Chem. 2021, 297, 100996. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, K.B.; Li, R. A prescription for ‘stress’—The role of Hsp90 in genome stability and cellular adaptation. Trends Cell Biol. 2012, 22, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.-H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef]
- Sanglard, D.; Ischer, F.; Parkinson, T.; Falconer, D.; Bille, J. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob. Agents Chemother. 2003, 47, 2404–2412. [Google Scholar] [CrossRef]
- Cowen, L.E.; Lindquist, S. Hsp90 potentiates the rapid evolution of new traits: Drug resistance in diverse fungi. Science 2005, 309, 2185–2189. [Google Scholar] [CrossRef]
- Robbins, N.; Leach, M.D.; Cowen, L.E. Lysine deacetylases Hda1 and Rpd3 regulate Hsp90 function thereby governing fungal drug resistance. Cell Rep. 2012, 2, 878–888. [Google Scholar] [CrossRef]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperon 2018, 23, 303–315. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward Primer | Reverse Primer |
|---|---|---|
| GAPDH | 5′-gaccgttgacggtccatcc-3′ | 5′-catcggtggttgggactc-3′ |
| CDR1 | 5′-aaagatgacctcgtcagcaggttt-3′ | 5′-ccaattcccaatttcgaaggt-3′ |
| CDR2 | 5′-tgttggtaccatttcatatttctgttg-3′ | 5′-aagagattgccaattgtcccata-3′ |
| MDR1 | 5′-tcgttttagcaatggcgtttg-3′ | 5′-ccatgccctccaatgaacag-3′ |
| ERG3 | 5′-tccagttgatgggttcttcc-3′ | 5′-ggacagtgtgacaagcgg-3′ |
| ERG11 | 5′-ttacctcattattggagacgtgatg-3′ | 5′-cacgttctcttctcagtttaatttctttc-3′ |
| HSP90 | 5′-aagtgctggtgctgacg-3′ | 5′-cttaccaccagcgttag-3′ |
| HSP30 | 5′-catgctccaactgctac-3′ | 5′-cgttcttcagcttcggc-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utkalaja, B.G.; Sahu, S.R.; Parida, S.S.; Acharya, N. Hsp90-Mediated Multi-Drug Resistance in DNA Polymerase-Defective Strains of Candida albicans. J. Fungi 2024, 10, 222. https://doi.org/10.3390/jof10030222
Utkalaja BG, Sahu SR, Parida SS, Acharya N. Hsp90-Mediated Multi-Drug Resistance in DNA Polymerase-Defective Strains of Candida albicans. Journal of Fungi. 2024; 10(3):222. https://doi.org/10.3390/jof10030222
Chicago/Turabian StyleUtkalaja, Bhabasha Gyanadeep, Satya Ranjan Sahu, Sushree Subhashree Parida, and Narottam Acharya. 2024. "Hsp90-Mediated Multi-Drug Resistance in DNA Polymerase-Defective Strains of Candida albicans" Journal of Fungi 10, no. 3: 222. https://doi.org/10.3390/jof10030222