
Epigenetics and Congenital Heart Diseases



Abstract
:1. Introduction
2. Morphogenesis, Embryology and Disease Spectrum
3. Epigenetic Alterations in Heart Disease
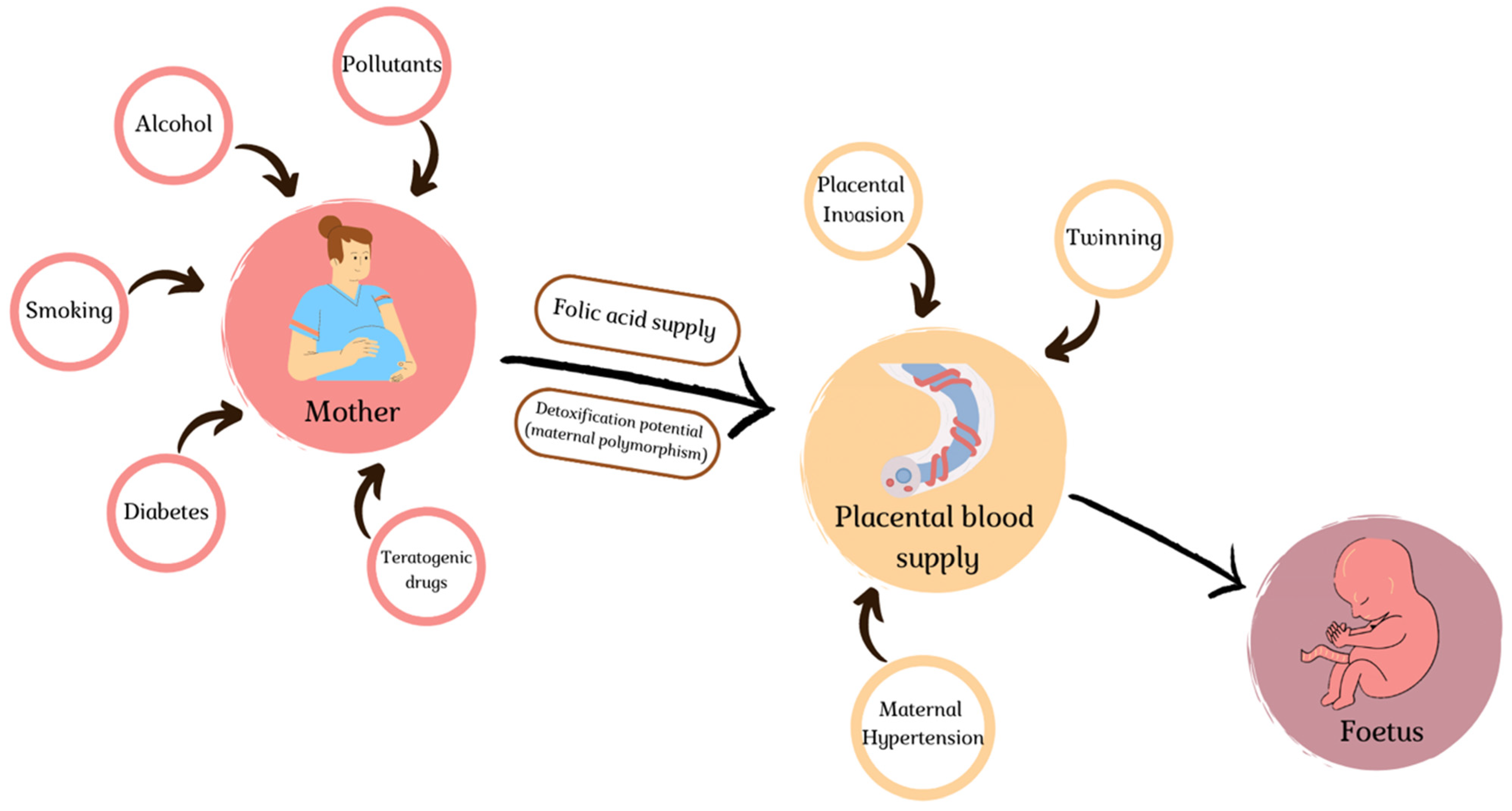
4. Environmental Slights
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- van der Linde, D.; Konings, E.E.M.; Slager, M.A.; Witsenburg, M.; Helbing, W.A.; Takkenberg, J.J.M.; Roos-Hesselink, J.W. Birth Prevalence of Congenital Heart Disease Worldwide: A Systematic Review and Meta-Analysis. J. Am. Coll. Cardiol. 2011, 58, 2241–2247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiene, G.; Frescura, C. Anatomical and Pathophysiological Classification of Congenital Heart Disease. Cardiovasc. Pathol. 2010, 19, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Bajolle, F.; Zaffran, S.; Bonnet, D. Genetics and Embryological Mechanisms of Congenital Heart Diseases. Arch. Cardiovasc. Dis. 2009, 102, 59–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, T.B.; Foo, S.Y.R.; Chen, C.K. The Role of Epigenetics in Congenital Heart Disease. Genes 2021, 12, 390. [Google Scholar] [CrossRef]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of Congenital Heart Disease: The Glass Half Empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De Novo Mutations in Histone-Modifying Genes in Congenital Heart Disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Imany-Shakibai, H.; Yin, O.; Russell, M.R.; Sklansky, M.; Satou, G.; Afshar, Y. Discordant Congenital Heart Defects in Monochorionic Twins: Risk Factors and Proposed Pathophysiology. PLoS ONE 2021, 16, e0251160. [Google Scholar] [CrossRef]
- AlRais, F.; Feldstein, V.A.; Srivastava, D.; Gosnell, K.; Moon-Grady, A.J. Monochorionic Twins Discordant for Congenital Heart Disease: A Referral Center’s Experience and Possible Pathophysiologic Mechanisms. Prenat. Diagn. 2011, 31, 978–984. [Google Scholar] [CrossRef]
- Sedmera, D. Function and Form in the Developing Cardiovascular System. Cardiovasc. Res. 2011, 91, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Srivastava, D. Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the Mammalian Heart from Two Sources of Myocardial Cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Restivo, A.; Piacentini, G.; Placidi, S.; Saffirio, C.; Marino, B. Cardiac Outflow Tract: A Review of Some Embryogenetic Aspects of the Conotruncal Region of the Heart. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Schleich, J.-M.; Abdulla, T.; Summers, R.; Houyel, L. An Overview of Cardiac Morphogenesis. Arch. Cardiovasc. Dis. 2013, 106, 612–623. [Google Scholar] [CrossRef]
- Sadler, T.W. Establishing the Embryonic Axes: Prime Time for Teratogenic Insults. J. Cardiovasc. Dev. Dis. 2017, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Gittenberger-de Groot, A.C.; Calkoen, E.E.; Poelmann, R.E.; Bartelings, M.M.; Jongbloed, M.R.M. Morphogenesis and Molecular Considerations on Congenital Cardiac Septal Defects. Ann. Med. 2014, 46, 640–652. [Google Scholar] [CrossRef]
- Azhar, M.; Ware, S.M. Genetic and Developmental Basis of Cardiovascular Malformations. Clin. Perinatol. 2016, 43, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Muntean, I.; Togănel, R.; Benedek, T. Genetics of Congenital Heart Disease: Past and Present. Biochem. Genet. 2017, 55, 105–123. [Google Scholar] [CrossRef]
- VanOudenhove, J.; Yankee, T.N.; Wilderman, A.; Cotney, J. Epigenomic and Transcriptomic Dynamics During Human Heart Organogenesis. Circ. Res. 2020, 127, e184–e209. [Google Scholar] [CrossRef]
- Acemel, R.D.; Maeso, I.; Gómez-Skarmeta, J.L. Topologically Associated Domains: A Successful Scaffold for the Evolution of Gene Regulation in Animals. Wiley Interdiscip. Rev. Dev. Biol. 2017, 6, e265. [Google Scholar] [CrossRef] [Green Version]
- George, R.M.; Firulli, A.B. Epigenetics and Heart Development. Front. Cell Dev. Biol. 2021, 9, 637996. [Google Scholar] [CrossRef] [PubMed]
- Piché, J.; Van Vliet, P.P.; Pucéat, M.; Andelfinger, G. The Expanding Phenotypes of Cohesinopathies: One Ring to Rule Them All! Cell Cycle Georget. Tex 2019, 18, 2828–2848. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone Core Modifications Regulating Nucleosome Structure and Dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Miller, S.A.; Weinmann, A.S. An Essential Interaction between T-Box Proteins and Histone-Modifying Enzymes. Epigenetics 2009, 4, 85–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulcoli, F.G.; Franzese, M.; Liu, X.; Zhang, Z.; Angelini, C.; Baldini, A. Rebalancing Gene Haploinsufficiency in Vivo by Targeting Chromatin. Nat. Commun. 2016, 7, 11688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.-S.; Wang, J.-Z.; Shi, S.; Han, Y.; Zhang, Y.; Zhi, J.-X.; Xu, C.; Li, F.-F.; Wang, G.-Y.; Liu, S.-L. Identification of Epigenetic Factor KAT2B Gene Variants for Possible Roles in Congenital Heart Diseases. Biosci. Rep. 2020, 40, BSR20191779. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Subrahmanyan, L.; Smith, E.; Yu, X.; Zaidi, S.; Choi, M.; Mane, S.; Nelson-Williams, C.; Behjati, M.; Kazemi, M.; et al. Mutations in the Histone Modifier PRDM6 Are Associated with Isolated Nonsyndromic Patent Ductus Arteriosus. Am. J. Hum. Genet. 2016, 98, 1082–1091. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Xiong, Y.; Dong, X.; Wang, H.; Qian, Y.; Ma, D.; Li, X. Genome-Wide Methylation Analysis Reveals Differentially Methylated CpG Sites and Altered Expression of Heart Development-Associated Genes in Fetuses with Cardiac Defects. Exp. Ther. Med. 2021, 22, 1032. [Google Scholar] [CrossRef]
- Chang, S.; Wang, Y.; Xin, Y.; Wang, S.; Luo, Y.; Wang, L.; Zhang, H.; Li, J. DNA Methylation Abnormalities of Imprinted Genes in Congenital Heart Disease: A Pilot Study. BMC Med. Genom. 2021, 14, 4. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Yilmaz, A.; Saiyed, N.M.; Mishra, N.K.; Guda, C.; Radhakrishna, U. Precision Cardiovascular Medicine: Artificial Intelligence and Epigenetics for the Pathogenesis and Prediction of Coarctation in Neonates. J. Matern.-Fetal Neonatal Med. 2022, 35, 457–464. [Google Scholar] [CrossRef]
- Grunert, M.; Appelt, S.; Grossfeld, P.; Sperling, S.R. The Needle in the Haystack-Searching for Genetic and Epigenetic Differences in Monozygotic Twins Discordant for Tetralogy of Fallot. J. Cardiovasc. Dev. Dis. 2020, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ye, M.; Xu, H.; Gu, R.; Ma, X.; Chen, M.; Li, X.; Sheng, W.; Huang, G. Methylation Status of CpG Sites in the NOTCH4 Promoter Region Regulates NOTCH4 Expression in Patients with Tetralogy of Fallot. Mol. Med. Rep. 2020, 22, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zang, L.-Q.; Li, X.-K.; Shu, Q. An Epigenetic View of Developmental Diseases: New Targets, New Therapies. World J. Pediatr. WJP 2016, 12, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Banks, K.M.; Pan, H.; Verma, N.; Dixon, G.R.; Zhou, T.; Ding, B.; Elemento, O.; Chen, S.; Huangfu, D.; et al. Stage-Specific Regulation of DNA Methylation by TET Enzymes during Human Cardiac Differentiation. Cell Rep. 2021, 37, 110095. [Google Scholar] [CrossRef] [PubMed]
- George, M.R.; Duan, Q.; Nagle, A.; Kathiriya, I.S.; Huang, Y.; Rao, K.; Haldar, S.M.; Bruneau, B.G. Minimal in Vivo Requirements for Developmentally Regulated Cardiac Long Intergenic Non-Coding RNAs. Dev. Camb. Engl. 2019, 146, dev185314. [Google Scholar] [CrossRef]
- Haunschild, J.; Schellinger, I.N.; Barnard, S.J.; von Aspern, K.; Davierwala, P.; Misfeld, M.; Petroff, D.; Borger, M.A.; Etz, C.D. Bicuspid Aortic Valve Patients Show Specific Epigenetic Tissue Signature Increasing Extracellular Matrix Destruction. Interact. Cardiovasc. Thorac. Surg. 2019, 29, 937–943. [Google Scholar] [CrossRef]
- Patterson, D. Molecular Genetic Analysis of Down Syndrome. Hum. Genet. 2009, 126, 195–214. [Google Scholar] [CrossRef]
- Vilardell, M.; Rasche, A.; Thormann, A.; Maschke-Dutz, E.; Pérez-Jurado, L.A.; Lehrach, H.; Herwig, R. Meta-Analysis of Heterogeneous Down Syndrome Data Reveals Consistent Genome-Wide Dosage Effects Related to Neurological Processes. BMC Genom. 2011, 12, 229. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Olson, S.B.; Maslen, C.L. A Review of Recent Developments in Turner Syndrome Research. J. Cardiovasc. Dev. Dis. 2021, 8, 138. [Google Scholar] [CrossRef]
- Yan, S.; Lu, J.; Jiao, K. Epigenetic Regulation of Cardiac Neural Crest Cells. Front. Cell Dev. Biol. 2021, 9, 678954. [Google Scholar] [CrossRef]
- Rufaihah, A.J.; Chen, C.K.; Yap, C.H.; Mattar, C.N.Z. Mending a Broken Heart: In Vitro, in Vivo and in Silico Models of Congenital Heart Disease. Dis. Model. Mech. 2021, 14, dmm047522. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, S.; Gao, H.; Han, L.; Chu, X.; Sheng, Y.; Shou, W.; Wang, Y.; Liu, Y.; Wan, J.; et al. Genome-Wide Studies Reveal the Essential and Opposite Roles of ARID1A in Controlling Human Cardiogenesis and Neurogenesis from Pluripotent Stem Cells. Genome Biol. 2020, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; van Vliet, P.P.; Andelfinger, G.; Puceat, M. Role of Epigenetics in Cardiac Development and Congenital Diseases. Physiol. Rev. 2018, 98, 2453–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, K.; Lazar, H.P.; Kurolap, A.; Martinez, A.F.; Paperna, T.; Cohen, L.; Smeland, M.F.; Whalen, S.; Heide, S.; Keren, B.; et al. The CHD4-Related Syndrome: A Comprehensive Investigation of the Clinical Spectrum, Genotype-Phenotype Correlations, and Molecular Basis. Genet. Med. 2020, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Lavery, W.J.; Barski, A.; Wiley, S.; Schorry, E.K.; Lindsley, A.W. KMT2C/D COMPASS Complex-Associated Diseases [KCDCOM-ADs]: An Emerging Class of Congenital Regulopathies. Clin. Epigenetics 2020, 12, 10. [Google Scholar] [CrossRef]
- Froimchuk, E.; Jang, Y.; Ge, K. Histone H3 Lysine 4 Methyltransferase KMT2D. Gene 2017, 627, 337–342. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Tischer, A.; Zimmermann, M.T.; Kemppainen, J.L.; Sastry, S.; Knight Johnson, A.E.; Cousin, M.A.; Boczek, N.J.; Oliver, G.; Misra, V.K.; et al. A Novel Kleefstra Syndrome-Associated Variant That Affects the Conserved TPLX Motif within the Ankyrin Repeat of EHMT1 Leads to Abnormal Protein Folding. J. Biol. Chem. 2017, 292, 3866–3876. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Yamaza, T.; Lee, J.S.; Yu, J.; Wang, S.; Fan, G.; Shi, S.; Wang, C.-Y. BCOR Regulates Mesenchymal Stem Cell Function by Epigenetic Mechanisms. Nat. Cell Biol. 2009, 11, 1002–1009. [Google Scholar] [CrossRef]
- Davoody, A.; Chen, I.-P.; Nanda, R.; Uribe, F.; Reichenberger, E.J. Oculofaciocardiodental Syndrome: A Rare Case and Review of the Literature. Cleft Palate-Craniofac. J. 2012, 49, e55–e60. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.M.; Schwartz, C.E. MED12 Related Disorders. Am. J. Med. Genet. A. 2013, 161A, 2734–2740. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, C.E.; Tarpey, P.S.; Lubs, H.A.; Verloes, A.; May, M.M.; Risheg, H.; Friez, M.J.; Futreal, P.A.; Edkins, S.; Teague, J.; et al. The Original Lujan Syndrome Family Has a Novel Missense Mutation (p.N1007S) in the MED12 Gene. J. Med. Genet. 2007, 44, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herskind, A.M.; Almind Pedersen, D.; Christensen, K. Increased Prevalence of Congenital Heart Defects in Monozygotic and Dizygotic Twins. Circulation 2013, 128, 1182–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahtiyar, M.O.; Dulay, A.T.; Weeks, B.P.; Friedman, A.H.; Copel, J.A. Prevalence of Congenital Heart Defects in Monochorionic/Diamniotic Twin Gestations: A Systematic Literature Review. J. Ultrasound Med. 2007, 26, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Karatza, A.A.; Wolfenden, J.L.; Taylor, M.J.O.; Wee, L.; Fisk, N.M.; Gardiner, H.M. Influence of Twin-Twin Transfusion Syndrome on Fetal Cardiovascular Structure and Function: Prospective Case-Control Study of 136 Monochorionic Twin Pregnancies. Heart Br. Card. Soc. 2002, 88, 271–277. [Google Scholar] [CrossRef]
- Hidaka, N.; Tsukimori, K.; Chiba, Y.; Hara, T.; Wake, N. Monochorionic Twins in Which at Least One Fetus Has a Congenital Heart Disease with or without Twin-Twin Transfusion Syndrome. J. Perinat. Med. 2007, 35, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Manning, N. The Influence of Twinning on Cardiac Development. Early Hum. Dev. 2008, 84, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Pruetz, J.D.; Sklansky, M.; Detterich, J.; Korst, L.M.; Llanes, A.; Chmait, R.H. Twin-Twin Transfusion Syndrome Treated with Laser Surgery: Postnatal Prevalence of Congenital Heart Disease in Surviving Recipients and Donors. Prenat. Diagn. 2011, 31, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Hove, J.R.; Köster, R.W.; Forouhar, A.S.; Acevedo-Bolton, G.; Fraser, S.E.; Gharib, M. Intracardiac Fluid Forces Are an Essential Epigenetic Factor for Embryonic Cardiogenesis. Nature 2003, 421, 172–177. [Google Scholar] [CrossRef]
- Johnson, B.; Bark, D.; Van Herck, I.; Garrity, D.; Dasi, L.P. Altered Mechanical State in the Embryonic Heart Results in Time-Dependent Decreases in Cardiac Function. Biomech. Model. Mechanobiol. 2015, 14, 1379–1389. [Google Scholar] [CrossRef]
- Rugonyi, S. Genetic and Flow Anomalies in Congenital Heart Disease. AIMS Genet. 2016, 3, 157–166. [Google Scholar] [CrossRef]
- Santhanakrishnan, A.; Miller, L.A. Fluid Dynamics of Heart Development. Cell Biochem. Biophys. 2011, 61, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Jarrell, D.K.; Lennon, M.L.; Jacot, J.G. Epigenetics and Mechanobiology in Heart Development and Congenital Heart Disease. Diseases 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, N.; Archer, N. A Study to Determine the Incidence of Structural Congenital Heart Disease in Monochorionic Twins. Prenat. Diagn. 2006, 26, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, J.; Beke, A.; Marton, T.; Hruby, E.; Pete, B.; Papp, Z. Congenital Heart Diseases in Twin Pregnancies. Fetal Diagn. Ther. 2006, 21, 198–203. [Google Scholar] [CrossRef]
- Yuan, S.; Zaidi, S.; Brueckner, M. Congenital Heart Disease: Emerging Themes Linking Genetics and Development. Curr. Opin. Genet. Dev. 2013, 23, 352–359. [Google Scholar] [CrossRef] [Green Version]
- Giorgione, V.; Fesslova, V.; Boveri, S.; Candiani, M.; Khalil, A.; Cavoretto, P. Adverse Perinatal Outcome and Placental Abnormalities in Pregnancies with Major Fetal Congenital Heart Defects: A Retrospective Case-Control Study. Prenat. Diagn. 2020, 40, 1390–1397. [Google Scholar] [CrossRef]
- Maslen, C.L. Recent Advances in Placenta-Heart Interactions. Front. Physiol. 2018, 9, 735. [Google Scholar] [CrossRef] [Green Version]
- Andescavage, N.N.; Limperopoulos, C. Placental Abnormalities in Congenital Heart Disease. Transl. Pediatr. 2021, 10, 2148–2156. [Google Scholar] [CrossRef]
- Ozcan, T.; Kikano, S.; Plummer, S.; Strainic, J.; Ravishankar, S. The Association of Fetal Congenital Cardiac Defects and Placental Vascular Malperfusion. Pediatr. Dev. Pathol. 2021, 24, 187–192. [Google Scholar] [CrossRef]
- Courtney, J.A.; Cnota, J.F.; Jones, H.N. The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front. Physiol. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Bateman, B.T.; Huybrechts, K.F.; Fischer, M.A.; Seely, E.W.; Ecker, J.L.; Oberg, A.S.; Franklin, J.M.; Mogun, H.; Hernandez-Diaz, S. Chronic Hypertension in Pregnancy and the Risk of Congenital Malformations: A Cohort Study. Am. J. Obstet. Gynecol. 2015, 212, 337.E1–337.E14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, A.; Lee, L.J.; Mitchell, L.E.; Agopian, A.J. Maternal Hypertension During Pregnancy and the Risk of Congenital Heart Defects in Offspring: A Systematic Review and Meta-Analysis. Pediatr. Cardiol. 2015, 36, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Leirgul, E.; Brodwall, K.; Greve, G.; Vollset, S.E.; Holmstrøm, H.; Tell, G.S.; Øyen, N. Maternal Diabetes, Birth Weight, and Neonatal Risk of Congenital Heart Defects in Norway, 1994–2009. Obstet. Gynecol. 2016, 128, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.A.; Verheijen, P.M.; Copel, J.A.; Kleinman, C.S.; Wassink, S.; Visser, G.H.A.; Meijboom, E.-J. Congenital Heart Disease in Pregnancies Complicated by Maternal Diabetes Mellitus. An International Clinical Collaboration, Literature Review, and Meta-Analysis. Herz 2010, 35, 19–26. [Google Scholar] [CrossRef]
- Ding, Z.; Zhou, H.; McCauley, N.; Ko, G.; Zhang, K.K.; Xie, L. In Ovo Hyperglycemia Causes Congenital Limb Defects in Chicken Embryos via Disruption of Cell Proliferation and Apoptosis. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165955. [Google Scholar] [CrossRef]
- Gabbay-Benziv, R.; Reece, E.A.; Wang, F.; Yang, P. Birth Defects in Pregestational Diabetes: Defect Range, Glycemic Threshold and Pathogenesis. World J. Diabetes 2015, 6, 481–488. [Google Scholar] [CrossRef]
- Wang, F.; Wu, Y.; Quon, M.J.; Li, X.; Yang, P. ASK1 Mediates the Teratogenicity of Diabetes in the Developing Heart by Inducing ER Stress and Inhibiting Critical Factors Essential for Cardiac Development. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E487–E499. [Google Scholar] [CrossRef] [Green Version]
- Persson, M.; Razaz, N.; Edstedt Bonamy, A.-K.; Villamor, E.; Cnattingius, S. Maternal Overweight and Obesity and Risk of Congenital Heart Defects. J. Am. Coll. Cardiol. 2019, 73, 44–53. [Google Scholar] [CrossRef]
- Helle, E.; Priest, J.R. Maternal Obesity and Diabetes Mellitus as Risk Factors for Congenital Heart Disease in the Offspring. J. Am. Heart Assoc. 2020, 9, e011541. [Google Scholar] [CrossRef]
- Linask, K.K.; Huhta, J. Folate Protection from Congenital Heart Defects Linked with Canonical Wnt Signaling and Epigenetics. Curr. Opin. Pediatr. 2010, 22, 561–566. [Google Scholar] [CrossRef]
- Padmanabhan, N.; Jia, D.; Geary-Joo, C.; Wu, X.; Ferguson-Smith, A.C.; Fung, E.; Bieda, M.C.; Snyder, F.F.; Gravel, R.A.; Cross, J.C.; et al. Mutation in Folate Metabolism Causes Epigenetic Instability and Transgenerational Effects on Development. Cell 2013, 155, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Li, J.; Wei, C.; Lu, Q.; Tang, X.; Erickson, S.W.; MacLeod, S.L.; Hobbs, C.A. A Three-Way Interaction among Maternal and Fetal Variants Contributing to Congenital Heart Defects. Ann. Hum. Genet. 2016, 80, 20–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, J.A.; Groom, A.; Potter, C.; Coneyworth, L.J.; Ford, D.; Mathers, J.C.; Relton, C.L. Genetic and Non-Genetic Influences during Pregnancy on Infant Global and Site Specific DNA Methylation: Role for Folate Gene Variants and Vitamin B12. PLoS ONE 2012, 7, e33290. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.O.; Chellappan, S.; Kukshal, P. Exploring the Role of Maternal Nutritional Epigenetics in Congenital Heart Disease. Curr. Dev. Nutr. 2020, 4, nzaa166. [Google Scholar] [CrossRef]
- Ionescu-Ittu, R.; Marelli, A.J.; Mackie, A.S.; Pilote, L. Prevalence of Severe Congenital Heart Disease after Folic Acid Fortification of Grain Products: Time Trend Analysis in Quebec, Canada. BMJ 2009, 338, b1673. [Google Scholar] [CrossRef] [Green Version]
- Eckmann-Scholz, C.; von Kaisenberg, C.S.; Alkatout, I.; Jonat, W.; Rajabi-Wieckhorst, A. Pathologic Ultrasound Findings and Risk for Congenital Anomalies in Teenage Pregnancies. J. Matern.-Fetal Neonatal Med. 2012, 25, 1950–1952. [Google Scholar] [CrossRef]
- Khalil, A.; Tanos, R.; El-Hachem, N.; Kurban, M.; Bouvagnet, P.; Bitar, F.; Nemer, G. A HAND to TBX5 Explains the Link Between Thalidomide and Cardiac Diseases. Sci. Rep. 2017, 7, 1416. [Google Scholar] [CrossRef] [Green Version]
- Gutgesell, H. Lithium Use in Pregnancy and the Risk of Cardiac Malformations. N. Engl. J. Med. 2017, 377, 893–894. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Serrano, M.C.; Lastra-Vicente, R.; Brinez, P.; Acharya, G.; Huhta, J.C.; Chen, R.; Linask, K.K. Folate Rescues Lithium-, Homocysteine- and Wnt3A-Induced Vertebrate Cardiac Anomalies. Dis. Model. Mech. 2009, 2, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Tanoshima, M.; Kobayashi, T.; Tanoshima, R.; Beyene, J.; Koren, G.; Ito, S. Risks of Congenital Malformations in Offspring Exposed to Valproic Acid in Utero: A Systematic Review and Cumulative Meta-Analysis. Clin. Pharmacol. Ther. 2015, 98, 417–441. [Google Scholar] [CrossRef]
- Duan, H.-Y.; Zhou, K.-Y.; Wang, T.; Zhang, Y.; Li, Y.-F.; Hua, Y.-M.; Wang, C. Disruption of Planar Cell Polarity Pathway Attributable to Valproic Acid-Induced Congenital Heart Disease through Hdac3 Participation in Mice. Chin. Med. J. 2018, 131, 2080–2088. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-H.; Ho, Y.-L.; Huang, P.-T.; Chu, S.-L.; Tsai, H.-J.; Liou, H.-H. The Phosphorylation State of GSK3β Serine 9 Correlated to the Development of Valproic Acid-Associated Fetal Cardiac Teratogenicity, Fetal VPA Syndrome, Rescued by Folic Acid Administration. Cardiovasc. Toxicol. 2016, 16, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, P.T.; Manziello, A.; Howard, J.; Palbykin, B.; Runyan, R.B.; Selmin, O. Gene Expression Profiling in the Fetal Cardiac Tissue after Folate and Low-Dose Trichloroethylene Exposure. Birt. Defects Res. A Clin. Mol. Teratol. 2010, 88, 111–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoll, R. Environmental Contaminants and Congenital Heart Defects: A Re-Evaluation of the Evidence. Int. J. Environ. Res. Public. Health 2018, 15, 2096. [Google Scholar] [CrossRef] [Green Version]
- Kuehl, K.S.; Loffredo, C.A. A Cluster of Hypoplastic Left Heart Malformation in Baltimore, Maryland. Pediatr. Cardiol. 2006, 27, 25–31. [Google Scholar] [CrossRef]
- de Gannes, M.; Ko, C.-I.; Zhang, X.; Biesiada, J.; Niu, L.; Koch, S.E.; Medvedovic, M.; Rubinstein, J.; Puga, A. Dioxin Disrupts Dynamic DNA Methylation Patterns in Genes That Govern Cardiomyocyte Maturation. Toxicol. Sci. 2020, 178, 325–337. [Google Scholar] [CrossRef]
- Vecoli, C.; Pulignani, S.; Andreassi, M.G. Genetic and Epigenetic Mechanisms Linking Air Pollution and Congenital Heart Disease. J. Cardiovasc. Dev. Dis. 2016, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Zhou, R.; Feng, Y.; Wang, Y. Mainstream Smoke and Sidestream Smoke Affect the Cardiac Differentiation of Mouse Embryonic Stem Cells Discriminately. Toxicology 2016, 357–358, 1–10. [Google Scholar] [CrossRef]
- Hoyt, A.T.; Canfield, M.A.; Romitti, P.A.; Botto, L.D.; Anderka, M.T.; Krikov, S.V.; Tarpey, M.K.; Feldkamp, M.L. Associations between Maternal Periconceptional Exposure to Secondhand Tobacco Smoke and Major Birth Defects. Am. J. Obstet. Gynecol. 2016, 215, 613.e1–613.e11. [Google Scholar] [CrossRef] [Green Version]
- Gianicolo, E.A.L.; Cresci, M.; Ait-Ali, L.; Foffa, I.; Andreassi, M.G. Smoking and Congenital Heart Disease: The Epidemiological and Biological Link. Curr. Pharm. Des. 2010, 16, 2572–2577. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, L.; Yang, T.; Chen, L.; Zhao, L.; Wang, T.; Chen, L.; Ye, Z.; Zheng, Z.; Qin, J. Parental Alcohol Consumption and the Risk of Congenital Heart Diseases in Offspring: An Updated Systematic Review and Meta-Analysis. Eur. J. Prev. Cardiol. 2020, 27, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, S.; Strainic, J.P.; Kim, J.; Ford, M.R.; Thrane, L.; Karunamuni, G.H.; Sheehan, M.M.; Chowdhury, A.; Gillespie, C.A.; Rollins, A.M.; et al. Glutathione Protects the Developing Heart from Defects and Global DNA Hypomethylation Induced by Prenatal Alcohol Exposure. Alcohol. Clin. Exp. Res. 2021, 45, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Zhu, J.; Lv, T.; Sun, H.; Huang, X.; Tian, J. Alcohol Consumption during Gestation Causes Histone3 Lysine9 Hyperacetylation and an Alternation of Expression of Heart Development-Related Genes in Mice. Alcohol. Clin. Exp. Res. 2014, 38, 2396–2402. [Google Scholar] [CrossRef]
- Chen, Z.; Li, S.; Guo, L.; Peng, X.; Liu, Y. Prenatal Alcohol Exposure Induced Congenital Heart Diseases: From Bench to Bedside. Birth Defects Res. 2021, 113, 521–534. [Google Scholar] [CrossRef]
- Tararbit, K.; Houyel, L.; Bonnet, D.; De Vigan, C.; Lelong, N.; Goffinet, F.; Khoshnood, B. Risk of Congenital Heart Defects Associated with Assisted Reproductive Technologies: A Population-Based Evaluation. Eur. Heart J. 2011, 32, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgione, V.; Parazzini, F.; Fesslova, V.; Cipriani, S.; Candiani, M.; Inversetti, A.; Sigismondi, C.; Tiberio, F.; Cavoretto, P. Congenital Heart Defects in IVF/ICSI Pregnancy: Systematic Review and Meta-Analysis. Ultrasound Obstet. Gynecol. 2018, 51, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panagiotopoulou, O.; Fouzas, S.; Sinopidis, X.; Mantagos, S.P.; Dimitriou, G.; Karatza, A.A. Congenital Heart Disease in Twins: The Contribution of Type of Conception and Chorionicity. Int. J. Cardiol. 2016, 218, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, J.; Romanelli, V.; Martin-Trujillo, A.; Fernández, G.-M.; Segovia, M.; Perandones, C.; Pérez Jurado, L.A.; Esteller, M.; Fraga, M.; Arias, P.; et al. Clinical and Molecular Analyses of Beckwith-Wiedemann Syndrome: Comparison between Spontaneous Conception and Assisted Reproduction Techniques. Am. J. Med. Genet. A. 2016, 170, 2740–2749. [Google Scholar] [CrossRef] [Green Version]
- Benincasa, G.; Marfella, R.; Della Mura, N.; Schiano, C.; Napoli, C. Strengths and Opportunities of Network Medicine in Cardiovascular Diseases. Circ. J. 2020, 84, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; Benincasa, G.; Donatelli, F.; Ambrosio, G. Precision Medicine in Distinct Heart Failure Phenotypes: Focus on Clinical Epigenetics. Am. Heart J. 2020, 224, 113–128. [Google Scholar] [CrossRef]
- Cheng, F.; Zhao, J.; Wang, Y.; Lu, W.; Liu, Z.; Zhou, Y.; Martin, W.R.; Wang, R.; Huang, J.; Hao, T.; et al. Comprehensive Characterization of Protein–Protein Interactions Perturbed by Disease Mutations. Nat. Genet. 2021, 53, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Springett, A.L.; Greenlees, R.; Loane, M.; Addor, M.-C.; Arriola, L.; Barisic, I.; Bergman, J.E.H.; Csaky-Szunyogh, M.; Dias, C.; et al. Trends in Congenital Anomalies in Europe from 1980 to 2012. PLoS ONE 2018, 13, e0194986. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Epigenetic Function | Syndrome Affected Gene | Cardiac Manifestation | Reference |
|---|---|---|---|
| ATP-dependent chromatin modifiers | Coffin–Siris syndrome ARID1A/B, SMARCB1, SMARCA4, SAMRCE1 | ASD VSD TOF PDA | [40,41,42] |
| CHARGE syndrome CHD7 | TOF DORV VSD AVSD PDA PS IAA | [40,43] | |
| Sifrim-Hitz-Weiss syndrome CHD4 | ASD VSD PS PDA TOF MV anomalies | [44] | |
| Williams syndrome WSTF | AS PS | [40] | |
| Histone modifiers | Kabuki syndrome KMT2D, KDM6A, WDR5 | Coarctation ASD VSD | [40,45,46] |
| Kleefstra syndrome EHMT1 | ASD VSD TOF Coarctation BAV PS | [40,45,47] | |
| Wolf–Hirschhorn syndrome WHSC1/2, LETM1 | ASD | [40] | |
| Rubinstein–Taybi CREBBP, EP300 | PDA ASD VSD HLHS BAV | [41,45] | |
| KAT2B | ASD VSD PDA TOF PS | [26] | |
| PRDM6 | PDA | [27] | |
| Oculofaciocardiodental syndrome BCOR | ASD VSD MV anomalies | [48,49] | |
| Cohesinopathies | Cornelia de Lange NIPBL, HDAC8, SMC1, SMC3, RAD21, BRD4, ANKRD11 | TOF ASD VSD PDA PS | [22,41] |
| Robert’s syndrome ESCO2 | VSD ASD PDA | [22] | |
| Warsaw breakage syndrome DDX11 | TOF VSD | [22] | |
| ARTX syndrome ATRX | VSD ASD TOF PDA PS/AS | [22] | |
| CHOPS syndrome AFF4 | VSD PDA | [22] | |
| STAG2-related X-linked Intellectual Deficiency STAG2 | VSD | [22] | |
| CAID syndrome SGO1 | PS/AS VSD | [22] | |
| Mediatorpathies | Opitz–Kaveggia syndrome MED12 | TOF | [43,50] |
| Lujan–Fryns syndrome MED12 | TOF | [43,51] | |
| Ohdo syndrome MED13L | TOF | [43] | |
| DNA methylation modulators | ICF syndrome DMNT3B | ASD VSD | [40] |
| Chromatin-modifier regulators | DiGeorge syndrome TBX1 | IAA Truncus arteriosus TOF TGA VSD | [21,43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linglart, L.; Bonnet, D. Epigenetics and Congenital Heart Diseases. J. Cardiovasc. Dev. Dis. 2022, 9, 185. https://doi.org/10.3390/jcdd9060185
Linglart L, Bonnet D. Epigenetics and Congenital Heart Diseases. Journal of Cardiovascular Development and Disease. 2022; 9(6):185. https://doi.org/10.3390/jcdd9060185
Chicago/Turabian StyleLinglart, Léa, and Damien Bonnet. 2022. "Epigenetics and Congenital Heart Diseases" Journal of Cardiovascular Development and Disease 9, no. 6: 185. https://doi.org/10.3390/jcdd9060185