
Endocardial Regulation of Cardiac Development

 and
and
Abstract
:1. Introduction
2. Development of the Endocardium
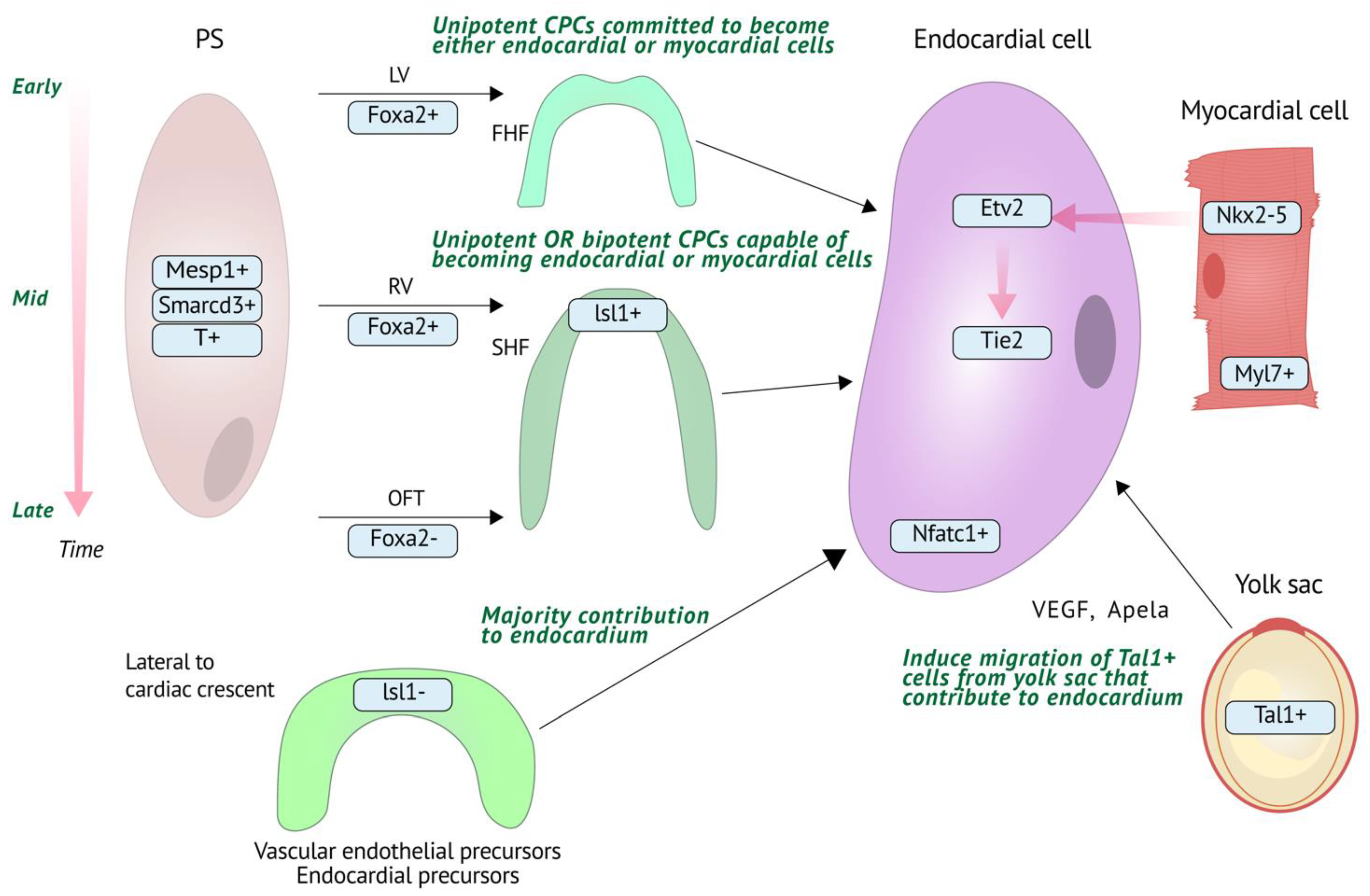
2.1. Endocardial Cell Origins
2.2. Molecular Regulation of Endocardial Development
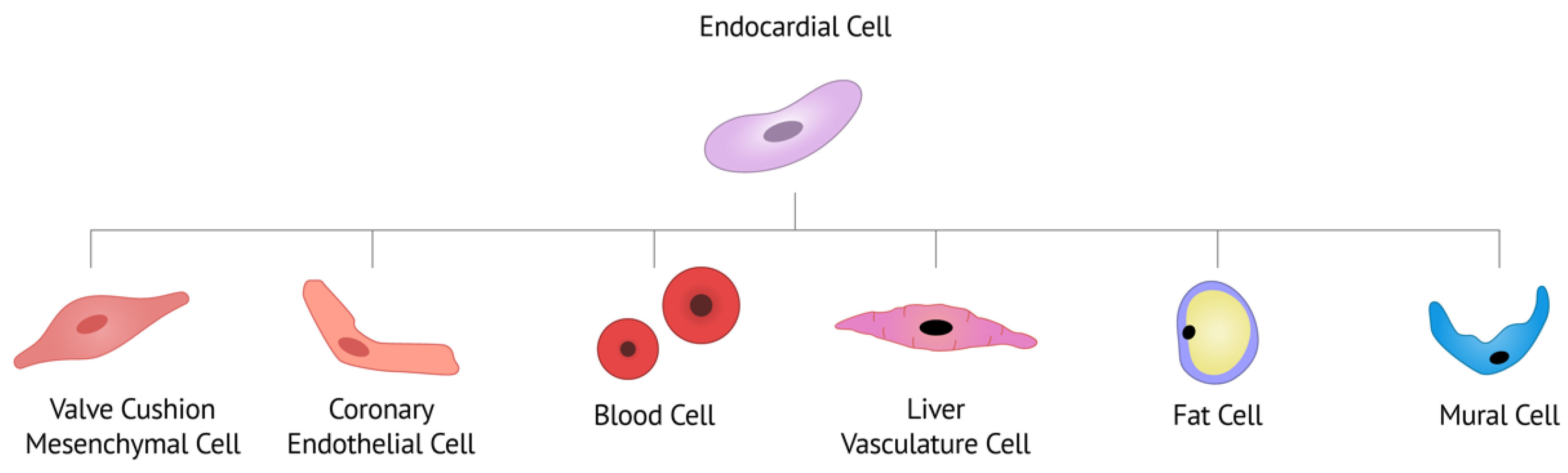
3. Endocardial Contribution to Cardiac Cell Types and Lineage-Specific Mechanisms
3.1. Hemogenic Endocardium
3.2. Cardiac Valves
3.3. Coronary Artery Vasculature
3.4. Liver Vasculature
3.5. Fat Cells
3.6. Mural Cells
4. Key Endocardial-Myocardial Molecular Pathways
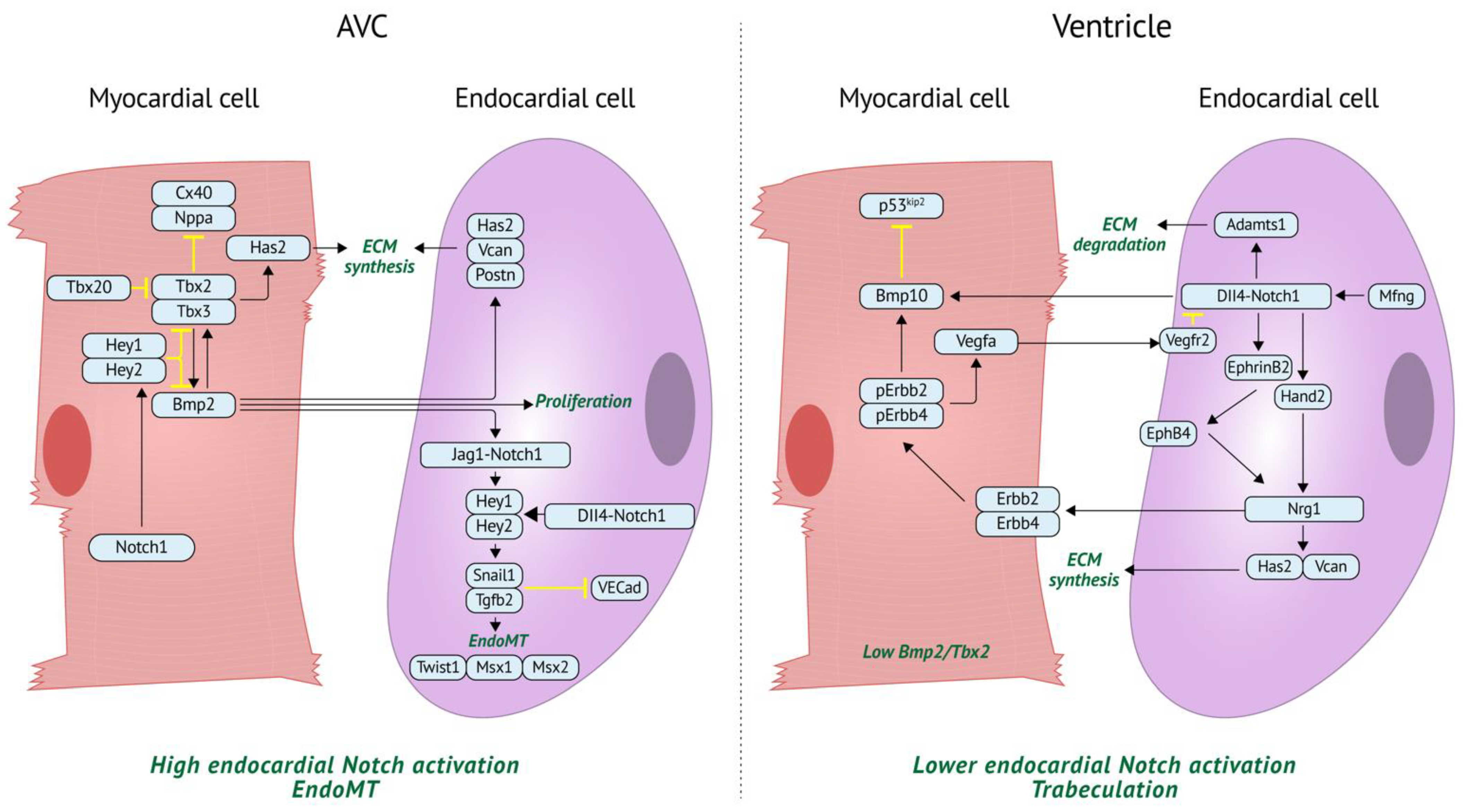
4.1. BMP Regulation of Myocardial Identity and Patterning
4.2. BMP Regulation of Valve Development
4.3. BMP Regulation of Trabeculation
4.4. Notch Regulation of Valve Development
4.5. Notch Regulation of Trabeculation
5. Endocardial Regulation of Other Cell Types/Processes
5.1. Additional Regulation of Valvulogenesis
5.2. Angiopoietin Regulation of Trabeculation and ECM Metabolism
5.3. Conduction System Development
6. The Role of Endocardium in Cardiac Disease
6.1. Bicuspid Aortic Valve
6.2. Mitral Valve Prolapse
6.3. Hypoplastic Left Heart Syndrome
7. Modeling Endocardial Development and Disease in Stem Cell-Derived Models and Organoids
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meilhac, S.M.; Buckingham, M.E. The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, M.; Wirrig, E.; Phelps, A.; Wessels, A. Extracellular matrix and heart development. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Nakaoka, Y.; Augustin, H.G.; Koh, G.Y. Myocardial Angiopoietin-1 Controls Atrial Chamber Morphogenesis by Spatiotemporal Degradation of Cardiac Jelly. Cell Rep. 2018, 23, 2455–2466. [Google Scholar] [CrossRef] [Green Version]
- Markwald, R.R.; Fitzharris, T.P.; Smith, W.N. Structural analysis of endocardial cytodifferentiation. Dev. Biol. 1975, 42, 160–180. [Google Scholar] [CrossRef]
- Markwald, R.R.; Fitzharris, T.P.; Manasek, F.J. Structural development of endocardial cushions. Am. J. Anat. 1977, 148, 85–119. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Gould, L.; Mikawa, T. The Fate Diversity of Mesodermal Cells within the Heart Field during Chicken Early Embryogenesis. Dev. Biol. 1996, 177, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Mikawa, T. Fate diversity of primitive streak cells during heart field formation in ovo. Dev. Dyn. 2000, 219, 505–513. [Google Scholar] [CrossRef]
- Bussmann, J.; Bakkers, J.; Schulte-Merker, S. Early Endocardial Morphogenesis Requires Scl/Tal1. PLoS Genet. 2007, 3, e140. [Google Scholar] [CrossRef]
- Schoenebeck, J.J.; Keegan, B.R.; Yelon, D. Vessel and Blood Specification Override Cardiac Potential in Anterior Mesoderm. Dev. Cell 2007, 13, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef] [Green Version]
- Masino, A.M.; Gallardo, T.D.; Wilcox, C.A.; Olson, E.N.; Williams, R.S.; Garry, D.J. Transcriptional Regulation of Cardiac Progenitor Cell Populations. Circ. Res. 2004, 95, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzi, M.P.; McCulley, D.J.; De Val, S.; Dodou, E.; Black, B.L. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol. 2005, 287, 134–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, A.; Caron, L.; Nakano, A.; Lam, J.T.; Bernshausen, A.; Chen, Y.; Qyang, Y.; Bu, L.; Sasaki, M.; Martin-Puig, S.; et al. Multipotent Embryonic Isl1+ Progenitor Cells Lead to Cardiac, Smooth Muscle, and Endothelial Cell Diversification. Cell 2006, 127, 1151–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattman, S.J.; Huber, T.L.; Keller, G.M. Multipotent Flk-1+ Cardiovascular Progenitor Cells Give Rise to the Cardiomyocyte, Endothelial, and Vascular Smooth Muscle Lineages. Dev. Cell 2006, 11, 723–732. [Google Scholar] [CrossRef] [Green Version]
- Misfeldt, A.M.; Boyle, S.C.; Tompkins, K.L.; Bautch, V.L.; Labosky, P.A.; Baldwin, H.S. Endocardial cells are a distinct endothelial lineage derived from Flk1+ multipotent cardiovascular progenitors. Dev. Biol. 2009, 333, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Collart, C.; Ciccarelli, A.; Ivanovitch, K.; Rosewell, I.; Kumar, S.; Kelly, G.; Edwards, A.; Smith, J.C. The migratory pathways of the cells that form the endocardium, dorsal aortae, and head vasculature in the mouse embryo. BMC Dev. Biol. 2021, 21, 8. [Google Scholar] [CrossRef]
- Milgrom-Hoffman, M.; Harrelson, Z.; Ferrara, N.; Zelzer, E.; Evans, S.M.; Tzahor, E. The heart endocardium is derived from vascular endothelial progenitors. Development 2011, 138, 4777–4787. [Google Scholar] [CrossRef] [Green Version]
- Devine, W.P.; Wythe, J.; George, M.; Koshiba-Takeuchi, K.; Bruneau, B.G. Early patterning and specification of cardiac progenitors in gastrulating mesoderm. eLife 2014, 3, e03848. [Google Scholar] [CrossRef]
- Meilhac, S.; Esner, M.; Kelly, R.; Nicolas, J.-F.; Buckingham, M.E. The Clonal Origin of Myocardial Cells in Different Regions of the Embryonic Mouse Heart. Dev. Cell 2004, 6, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Ivanovitch, K.; Soro-Barrio, P.; Chakravarty, P.; Jones, R.A.; Bell, D.M.; Gharavy, S.N.M.; Stamataki, D.; Delile, J.; Smith, J.C.; Briscoe, J. Ventricular, atrial, and outflow tract heart progenitors arise from spatially and molecularly distinct regions of the primitive streak. PLoS Biol. 2021, 19, e3001200. [Google Scholar] [CrossRef]
- Harris, I.S.; Black, B.L. Development of the Endocardium. Pediatr. Cardiol. 2010, 31, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferdous, A.; Caprioli, A.; Iacovino, M.; Martin, C.M.; Morris, J.; Richardson, J.A.; Latif, S.; Hammer, R.E.; Harvey, R.P.; Olson, E.N.; et al. Nkx2–5 transactivates the Ets-related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc. Natl. Acad. Sci. USA 2009, 106, 814–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saba, R.; Kitajima, K.; Rainbow, L.; Engert, S.; Uemura, M.; Ishida, H.; Kokkinopoulos, I.; Shintani, Y.; Miyagawa, S.; Kanai, Y.; et al. Endocardium differentiation through Sox17 expression in endocardium precursor cells regulates heart development in mice. Sci. Rep. 2019, 9, 11953. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.G.; Biben, C.; Elefanty, A.; Barnett, L.; Koentgen, F.; Robb, L.; Harvey, R.P. Efficient Cre-Mediated Deletion in Cardiac Progenitor Cells Conferred by a 3′UTR-Ires-Cre Allele of the Homeobox Gene Nkx2–5. Int. J. Dev. Biol. 2002, 46, 431–439. [Google Scholar] [PubMed]
- Palencia-Desai, S.; Kohli, V.; Kang, J.; Chi, N.C.; Black, B.; Sumanas, S. Vascular endothelial and endocardial progenitors differentiate as cardiomyocytes in the absence of Etsrp/Etv2 function. Development 2011, 138, 4721–4732. [Google Scholar] [CrossRef] [Green Version]
- Palencia-Desai, S.; Rost, M.S.; Schumacher, J.A.; Ton, Q.V.; Craig, M.P.; Baltrunaite, K.; Koenig, A.; Wang, J.; Poss, K.; Chi, N.C.; et al. Myocardium and BMP signaling are required for endocardial differentiation. Development 2015, 142, 2304–2315. [Google Scholar] [CrossRef] [Green Version]
- Engert, S.; Liao, W.P.; Burtscher, I.; Lickert, H. Sox17-2A-iCre: A knock-in mouse line expressing Cre recombinase in endoderm and vascular endothelial cells. Genes 2009, 47, 603–610. [Google Scholar] [CrossRef]
- Liu, Y.; Asakura, M.; Inoue, H.; Nakamura, T.; Sano, M.; Niu, Z.; Chen, M.; Schwartz, R.J.; Schneider, M.D. Sox17 is essential for the specification of cardiac mesoderm in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 3859–3864. [Google Scholar] [CrossRef] [Green Version]
- Nakano, A.; Nakano, H.; Smith, K.A.; Palpant, N.J. The developmental origins and lineage contributions of endocardial endothelium. Biochim. Biophys. Acta 2016, 1863, 1937–1947. [Google Scholar] [CrossRef]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef] [PubMed]
- Zovein, A.C.; Hofmann, J.J.; Lynch, M.; French, W.J.; Turlo, K.A.; Yang, Y.; Becker, M.S.; Zanetta, L.; Dejana, E.; Gasson, J.C.; et al. Fate Tracing Reveals the Endothelial Origin of Hematopoietic Stem Cells. Cell Stem Cell 2008, 3, 625–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Handel, B.; Montel-Hagen, A.; Sasidharan, R.; Nakano, H.; Ferrari, R.; Boogerd, C.J.; Schredelseker, J.; Wang, Y.; Hunter, S.; Org, T.; et al. Scl Represses Cardiomyogenesis in Prospective Hemogenic Endothelium and Endocardium. Cell 2012, 150, 590–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scialdone, A.; Tanaka, Y.; Jawaid, W.; Moignard, V.R.; Wilson, N.K.; Macaulay, I.; Marioni, A.S.I.C.M.J.C.; Gottgens, B. Resolving early mesoderm diversification through single-cell expression profiling. Nature 2016, 535, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Nakano, H.; Liu, X.; Arshi, A.; Nakashima, Y.; van Handel, B.; Sasidharan, R.; Harmon, A.W.; Shin, J.-H.; Schwartz, R.J.; Conway, S.J.; et al. Haemogenic endocardium contributes to transient definitive haematopoiesis. Nat. Commun. 2013, 4, 1564. [Google Scholar] [CrossRef] [Green Version]
- Zamir, L.; Singh, R.; Nathan, E.; Patrick, R.; Yifa, O.; Yahalom-Ronen, Y.; Arraf, A.A.; Schultheiss, T.M.; Suo, S.; Han, J.-D.J.; et al. Nkx2.5 marks angioblasts that contribute to hemogenic endothelium of the endocardium and dorsal aorta. eLife 2017, 6, e20994. [Google Scholar] [CrossRef] [Green Version]
- Shigeta, A.; Huang, V.; Zuo, J.; Besada, R.; Nakashima, Y.; Lu, Y.; Ding, Y.; Pellegrini, M.; Kulkarni, R.P.; Hsiai, T.; et al. Endocardially Derived Macrophages Are Essential for Valvular Remodeling. Dev. Cell 2019, 48, 617–630.e3. [Google Scholar] [CrossRef] [Green Version]
- De Lange, F.J.; Moorman, A.F.M.; Anderson, R.H.; Männer, J.; Soufan, A.T.; Vries, C.D.G.-D.; Schneider, M.; Webb, S.; van den Hoff, M.J.B.; Christoffels, V.M. Lineage and Morphogenetic Analysis of the Cardiac Valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef]
- De La Cruz, M.V.; Giménez-Ribotta, M.; Saravalli, O.; Cayré, R. The contribution of the inferior endocardial cushion of the atrioventricular canal to cardiac septation and to the development of the atrioventricular valves: Study in the chick embryo. Am. J. Anat. 1983, 166, 63–72. [Google Scholar] [CrossRef]
- Oosthoek, P.W.; Wenink, A.C.G.; Vrolijk, B.C.M.; Wisse, L.J.; DeRuiter, M.C.; Poelmann, R.E.; Groot, A.C.G.-D. Development of the atrioventricular valve tension apparatus in the human heart. Anat. Embryol. 1998, 198, 317–329. [Google Scholar] [CrossRef]
- Jiang, X.; Rowitch, D.; Soriano, P.; McMahon, A.; Sucov, H. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Eley, L.; Alqahtani, A.M.; MacGrogan, D.; Richardson, R.V.; Murphy, L.; Salguero-Jiménez, A.; Pedro, M.S.R.S.; Tiurma, S.; McCutcheon, L.; Gilmore, A.; et al. A novel source of arterial valve cells linked to bicuspid aortic valve without raphe in mice. eLife 2018, 7, e34110. [Google Scholar] [CrossRef] [PubMed]
- Mifflin, J.J.; Dupuis, L.E.; Alcala, N.E.; Russell, L.G.; Kern, C.B. Intercalated cushion cells within the cardiac outflow tract are derived from the myocardial troponin T type 2 (Tnnt2) Cre lineage. Dev. Dyn. 2018, 247, 1005–1017. [Google Scholar] [CrossRef]
- Peterson, J.C.; Chughtai, M.; Wisse, L.J.; Groot, A.C.G.-D.; Feng, Q.; Goumans, M.-J.T.H.; Van Munsteren, J.C.; Jongbloed, M.R.M.; De Ruiter, M.C. Nos3 mutation leads to abnormal neural crest cell and second heart field lineage patterning in bicuspid aortic valve formation. Dis. Model. Mech. 2018, 11, 34637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groot, A.C.G.-D.; Peeters, M.-P.F.V.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-Derived Cells Contribute a Novel Population to the Myocardial Wall and the Atrioventricular Cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessels, A.; Hoff, M.J.V.D.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.I.; Sharma, B.; Akerberg, B.N.; Numi, H.J.; Kivelä, R.; Saharinen, P.; Aghajanian, H.; McKay, A.S.; Bogard, P.E.; Chang, A.H.; et al. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 2014, 141, 4500–4512. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Pu, W.T.; Zhou, B. Cellular Origin and Developmental Program of Coronary Angiogenesis. Circ. Res. 2015, 116, 515–530. [Google Scholar] [CrossRef]
- Sharma, B.; Ho, L.; Ford, G.H.; Chen, H.; Goldstone, A.B.; Woo, Y.J.; Quertermous, T.; Reversade, B.; Red-Horse, K. Alternative Progenitor Cells Compensate to Rebuild the Coronary Vasculature in Elabela- and Apj-Deficient Hearts. Dev. Cell 2017, 42, 655–666.e3. [Google Scholar] [CrossRef] [Green Version]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct Compartments of the Proepicardial Organ Give Rise to Coronary Vascular Endothelial Cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Red-Horse, K.; Ueno, H.; Weissman, I.L.; Krasnow, M.A. Coronary arteries form by developmental reprogramming of venous cells. Nature 2010, 464, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Zhang, Z.; Lui, W.; Chen, X.; Wang, Y.; Chamberlain, A.A.; Moreno-Rodriguez, R.A.; Markwald, R.R.; O’Rourke, B.P.; Sharp, D.J.; et al. Endocardial Cells Form the Coronary Arteries by Angiogenesis through Myocardial-Endocardial VEGF Signaling. Cell 2012, 151, 1083–1096. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Lui, K.; Zhou, B. The Formation of Coronary Vessels in Cardiac Development and Disease. Cold Spring Harb. Perspect. Biol. 2019, 12, a037168. [Google Scholar] [CrossRef]
- Zhang, H.; Pu, W.; Li, G.; Huang, X.; He, L.; Tian, X.; Liu, Q.; Zhang, L.; Wu, S.M.; Sucov, H.M.; et al. Endocardium Minimally Contributes to Coronary Endothelium in the Embryonic Ventricular Free Walls. Circ. Res. 2016, 118, 1880–1893. [Google Scholar] [CrossRef] [Green Version]
- Guimarães-Camboa, N.; Stowe, J.; Aneas, I.; Sakabe, N.; Cattaneo, P.; Henderson, L.; Kilberg, M.S.; Johnson, R.; Chen, J.; McCulloch, A.D.; et al. HIF1α Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev. Cell 2015, 33, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Zhu, H.; Tian, X.; Wang, H.; Liu, S.; Liu, K.; Zhao, H.; He, L.; Huang, X.; Feng, Z.; et al. Extension of Endocardium-Derived Vessels Generate Coronary Arteries in Neonates. Circ. Res. 2022, 130, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Zhang, H.; He, L.; Huang, X.; Li, Y.; Pu, W.; Yu, W.; Zhang, L.; Cai, D.; Lui, K.O.; et al. Genetic Fate Mapping Defines the Vascular Potential of Endocardial Cells in the Adult Heart. Circ. Res. 2018, 122, 984–993. [Google Scholar] [CrossRef]
- Zhang, H.; Pu, W.; Tian, X.; Huang, X.; He, L.; Liu, Q.; Lingjuan, H.; Zhang, L.; He, L.; Liu, K.; et al. Genetic lineage tracing identifies endocardial origin of liver vasculature. Nat. Genet. 2016, 48, 537–543. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Cavallero, S.; Patterson, M.; Shen, H.; Xu, J.; Kumar, S.R.; Sucov, H.M. Adipogenesis and epicardial adipose tissue: A novel fate of the epicardium induced by mesenchymal transformation and PPARγ activation. Proc. Natl. Acad. Sci. USA 2015, 112, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pu, W.; Liu, Q.; He, L.; Huang, X.; Tian, X.; Zhang, L.; Nie, Y.; Hu, S.; Lui, K.O.; et al. Endocardium Contributes to Cardiac Fat. Circ. Res. 2016, 118, 254–265. [Google Scholar] [CrossRef]
- Cattaneo, P.; Mukherjee, D.; Spinozzi, S.; Zhang, L.; Larcher, V.; Stallcup, W.B.; Kataoka, H.; Chen, J.; Dimmeler, S.; Evans, S.M.; et al. Parallel Lineage-Tracing Studies Establish Fibroblasts as the Prevailing In Vivo Adipocyte Progenitor. Cell Rep. 2020, 30, 571–582.e2. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.-L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; Von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Mellgren, A.M.; Smith, C.L.; Olsen, G.S.; Eskiocak, B.; Zhou, B.; Kazi, M.N.; Ruiz, F.R.; Pu, W.T.; Tallquist, M.D. Platelet-Derived Growth Factor Receptor β Signaling Is Required for Efficient Epicardial Cell Migration and Development of Two Distinct Coronary Vascular Smooth Muscle Cell Populations. Circ. Res. 2008, 103, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-Derived Cell Epithelial-to-Mesenchymal Transition and Fate Specification Require PDGF Receptor Signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, H.; Liu, Y.; Adams, S.; Eilken, H.; Stehling, M.; Corada, M.; Dejana, E.; Zhou, B.; Adams, R.H. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat. Commun. 2016, 7, 12422. [Google Scholar] [CrossRef]
- Huang, X.; Feng, T.; Jiang, Z.; Meng, J.; Kou, S.; Lu, Z.; Chen, W.; Lin, C.-P.; Zhou, B.; Zhang, H. Dual lineage tracing identifies intermediate mesenchymal stage for endocardial contribution to fibroblasts, coronary mural cells, and adipocytes. J. Biol. Chem. 2019, 294, 8894–8906. [Google Scholar] [CrossRef]
- Yamadaa, M.; Revellib, J.-P.; Eichelebc, G.; Barrona, M.; Schwartz, R.J. Expression of Chick Tbx-2, Tbx-3, and Tbx-5 Genes during Early Heart Development: Evidence for BMP2 Induction of Tbx2. Dev. Biol. 2000, 228, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Lu, M.-F.; Schwartz, R.J.; Martin, J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development 2005, 132, 5601–5611. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Feliciano, J.; Tabin, C.J. Bmp2 instructs cardiac progenitors to form the heart-valve-inducing field. Dev. Biol. 2006, 295, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Prados, B.; Gómez-Apiñániz, P.; Papoutsi, T.; Luxán, G.; Zaffran, S.; Pérez-Pomares, J.M.; De La Pompa, J.L. Myocardial Bmp2 gain causes ectopic EMT and promotes cardiomyocyte proliferation and immaturity. Cell Death Dis. 2018, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Harrelson, Z.; Kelly, R.; Goldin, S.N.; Gibson-Brown, J.J.; Bollag, R.J.; Silver, L.M.; Papaioannou, V. Tbx2 is essential for patterning the atrioventricular canal and for morphogenesis of the outflow tract during heart development. Development 2004, 131, 5041–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, M.; Imanaka-Yoshida, K.; Schneider, M.D.; Schwartz, R.J.; Morisaki, T. T-box 2, a mediator of Bmp-Smad signaling, induced hyaluronan synthase 2 and Tgfβ2 expression and endocardial cushion formation. Proc. Natl. Acad. Sci. USA 2009, 106, 18604–18609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.-L.; Zhou, W.; Yang, L.; Bu, L.; Qyang, Y.; Zhang, X.; Li, X.; Rosenfeld, M.G.; Chen, J.; Evans, S. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development 2005, 132, 2475–2487. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Horsthuis, T.; Farin, H.F.; Grieskamp, T.; Norden, J.; Petry, M.; Wakker, V.; Moorman, A.F.; Christoffels, V.M.; Kispert, A. Tbx20 Interacts with Smads to Confine Tbx2 Expression to the Atrioventricular Canal. Circ. Res. 2009, 105, 442–452. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, T.; Nakajima, Y.; Miyazono, K.; Nakamura, H. Bone Morphogenetic Protein-2 Acts Synergistically with Transforming Growth Factor-Beta3 during Endothelial-Mesenchymal Transformation in the Developing Chick Heart. J. Cell. Physiol. 1999, 180, 35–45. [Google Scholar] [CrossRef]
- Yamagishi, T.; Nakajima, Y.; Nakamura, H. Expression of TGFβ3 RNA during chick embryogenesis: A possible important role in cardiovascular development. Cell Tissue Res. 1999, 298, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Yamagishi, T.; Hokari, S.; Nakamura, H. Mechanisms Involved in Valvuloseptal Endocardial Cushion Formation in Early Cardiogenesis: Roles of Transforming Growth Factor (TGF)-Beta and Bone Morphogenetic Protein (BMP). Anat. Rec. 2000, 258, 119–127. [Google Scholar] [CrossRef]
- Sugi, Y.; Yamamura, H.; Okagawa, H.; Markwald, R.R. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev. Biol. 2004, 269, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Okagawa, H.; Markwald, R.R.; Sugi, Y. Functional BMP receptor in endocardial cells is required in atrioventricular cushion mesenchymal cell formation in chick. Dev. Biol. 2007, 306, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Hoogaars, W.M.; Barnett, P.; Grieskamp, T.; Rana, M.S.; Buermans, H.; Farin, H.F.; Petry, M.; Heallen, T.; Martin, J.F.; et al. Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell. Mol. Life Sci. 2012, 69, 1377–1389. [Google Scholar] [CrossRef] [Green Version]
- Inai, K.; Norris, R.A.; Hoffman, S.; Markwald, R.R.; Sugi, Y. BMP-2 induces cell migration and periostin expression during atrioventricular valvulogenesis. Dev. Biol. 2008, 315, 383–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inai, K.; Burnside, J.L.; Hoffman, S.; Toole, B.P.; Sugi, Y. BMP-2 Induces Versican and Hyaluronan That Contribute to Post-EMT AV Cushion Cell Migration. PLoS ONE 2013, 8, e77593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, R.A.; Moreno-Rodriguez, R.A.; Sugi, Y.; Hoffman, S.; Amos, J.; Hart, M.M.; Potts, J.D.; Goodwin, R.L.; Markwald, R.R. Periostin regulates atrioventricular valve maturation. Dev. Biol. 2008, 316, 200–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxon, J.G.; Baer, D.R.; Barton, J.A.; Hawkins, T.; Wu, B.; Trusk, T.C.; Harris, S.E.; Zhou, B.; Mishina, Y.; Sugi, Y. BMP2 expression in the endocardial lineage is required for AV endocardial cushion maturation and remodeling. Dev. Biol. 2017, 430, 113–128. [Google Scholar] [CrossRef]
- Jiao, K.; Kulessa, H.; Tompkins, K.; Zhou, Y.; Batts, L.; Baldwin, H.S.; Hogan, B.L. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003, 17, 2362–2367. [Google Scholar] [CrossRef] [Green Version]
- McCulley, D.J.; Kang, J.-O.; Martin, J.F.; Black, B.L. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dyn. 2008, 237, 3200–3209. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shi, S.; Acosta, L.; Li, W.; Lu, J.; Bao, S.; Chen, Z.; Yang, Z.; Schneider, M.D.; Chien, K.R.; et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 2004, 131, 2219–2231. [Google Scholar] [CrossRef] [Green Version]
- Walsh, E.C.; Stainier, D.Y.R. UDP-Glucose Dehydrogenase Required for Cardiac Valve Formation in Zebrafish. Science 2001, 293, 1670–1673. [Google Scholar] [CrossRef]
- Hurlstone, A.; Haramis, A.-P.G.; Wienholds, E.; Begthel, H.; Korving, J.; Van Eeden, F.; Cuppen, E.; Zivkovic, D.; Plasterk, R.H.A.; Clevers, H. The Wnt/β-catenin pathway regulates cardiac valve formation. Nature 2003, 425, 633–637. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2003, 18, 99–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokubo, H.; Miyagawa-Tomita, S.; Tomimatsu, H.; Nakashima, Y.; Nakazawa, M.; Saga, Y.; Johnson, R.L. Targeted Disruption of hesr2 Results in Atrioventricular Valve Anomalies That Lead to Heart Dysfunction. Circ. Res. 2004, 95, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokubo, H.; Miyagawa-Tomita, S.; Nakazawa, M.; Saga, Y.; Johnson, R.L. Mouse hesr1 and hesr2 genes are redundantly required to mediate Notch signaling in the developing cardiovascular system. Dev. Biol. 2005, 278, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Steidl, C.; Wagner, T.U.; Lang, E.; Jakob, P.M.; Friedl, P.; Knobeloch, K.-P.; Gessler, M. Combined Loss of Hey1 and HeyL Causes Congenital Heart Defects Because of Impaired Epithelial to Mesenchymal Transition. Circ. Res. 2007, 100, 856–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Zurita, L.; Prados, B.; Grego-Bessa, J.; Luxán, G.; del Monte, G.; Benguría, A.; Adams, R.H.; Pérez-Pomares, J.M.; de la Pompa, J.L. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J. Clin. Investig. 2010, 120, 3493–3507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papoutsi, T.; Luna-Zurita, L.; Prados, B.; Zaffran, S.; de la Pompa, J.L. Bmp2 and Notch cooperate to pattern the embryonic endocardium. Development 2018, 145, dev.163378. [Google Scholar] [CrossRef] [Green Version]
- MacGrogan, D.; D’Amato, G.; Travisano, S.; Martinez-Poveda, B.; Luxán, G.; del Monte-Nieto, G.; Papoutsi, T.; Sbroggio, M.; Bou, V.; Arco, P.G.-D.; et al. Sequential Ligand-Dependent Notch Signaling Activation Regulates Valve Primordium Formation and Morphogenesis. Circ. Res. 2016, 118, 1480–1497. [Google Scholar] [CrossRef]
- Torregrosa-Carrión, R.; Zurita, L.L.; García-Marqués, F.; D’Amato, G.; Piñeiro-Sabarís, R.; Bonzón-Kulichenko, E.; Vázquez, J.; de la Pompa, J.L. NOTCH Activation Promotes Valve Formation by Regulating the Endocardial Secretome. Mol. Cell. Proteom. 2019, 18, 1782–1795. [Google Scholar] [CrossRef]
- Meyer, D.; Birchmeier, C. Multiple essential functions of neuregulin in development. Nature 1995, 378, 386–390. [Google Scholar] [CrossRef]
- Lee, K.-F.; Simon, H.; Chen, H.; Bates, B.; Hung, M.-C.; Hauser, C. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 1995, 378, 394–398. [Google Scholar] [CrossRef]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.U.; Chen, Z.-F.; Anderson, D.J. Molecular Distinction and Angiogenic Interaction between Embryonic Arteries and Veins Revealed by ephrin-B2 and Its Receptor Eph-B4. Cell 1998, 93, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Grego-Bessa, J.; Zurita, L.L.; del Monte, G.; Bolós, V.; Melgar, P.; Arandilla, A.; Garratt, A.; Zang, H.; Mukouyama, Y.-S.; Chen, H.; et al. Notch Signaling Is Essential for Ventricular Chamber Development. Dev. Cell 2007, 12, 415–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Monte-Nieto, G.; Ramialison, M.; Adam, A.A.; Wu, B.; Aharonov, A.; D’Uva, G.; Bourke, L.; Pitulescu, M.E.; Chen, H.; De La Pompa, J.L.; et al. Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 2018, 557, 439–445. [Google Scholar] [CrossRef]
- VanDusen, N.J.; Casanovas, J.; Vincentz, J.W.; Firulli, B.A.; Osterwalder, M.; Lopez-Rios, J.; Zeller, R.; Zhou, B.; Grego-Bessa, J.; De La Pompa, J.L.; et al. Hand2 Is an Essential Regulator for Two Notch-Dependent Functions within the Embryonic Endocardium. Cell Rep. 2014, 9, 2071–2083. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, G.; Luxán, G.; Nieto, G.D.M.; Poveda, B.M.; Torroja, C.; Walter, W.; Bochter, M.S.; Benedito, R.; Cole, S.E.; Martinez, F.; et al. Sequential Notch activation regulates ventricular chamber development. Nat. Cell Biol. 2016, 18, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; von Gise, A.; Liu, Q.; Hu, T.; Tian, X.; He, L.; Pu, W.; Huang, X.; He, L.; Cai, C.-L.; et al. Yap1 Is Required for Endothelial to Mesenchymal Transition of the Atrioventricular Cushion. J. Biol. Chem. 2014, 289, 18681–18692. [Google Scholar] [CrossRef] [Green Version]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig. 2000, 106, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Camenisch, T.D.; Schroeder, J.A.; Bradley, J.; Klewer, S.E.; McDonald, J.A. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2–ErbB3 receptors. Nat. Med. 2002, 8, 850–855. [Google Scholar] [CrossRef]
- Chen, B.; Bronson, R.T.; Klaman, L.D.; Hampton, T.G.; Wang, J.-F.; Green, P.J.; Magnuson, T.; Douglas, P.S.; Morgan, J.P.; Neel, B.G. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat. Genet. 2000, 24, 296–299. [Google Scholar] [CrossRef]
- Rivera-Feliciano, J.; Lee, K.-H.; Kong, S.W.; Rajagopal, S.; Ma, Q.; Springer, Z.; Izumo, S.; Tabin, C.J.; Pu, W.T. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development 2006, 133, 3607–3618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, W.M.; Deng, J.M.; Zhang, Z.P.; Behringer, R.R.; De Crombrugghe, B. Sox9 is required for cartilage formation. Nat. Genet. 1999, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Chaboissier, M.-C.; Kobayashi, A.; Vidal, V.I.P.; Lützkendorf, S.; van de Kant, H.J.G.; Wegner, M.; de Rooij, D.; Behringer, R.R.; Schedl, A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 2004, 131, 1891–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoln, J.; Kist, R.; Scherer, G.; Yutzey, K.E. Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev. Biol. 2007, 305, 120–132. [Google Scholar] [CrossRef] [Green Version]
- De La Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [CrossRef]
- Wu, B.; Wang, Y.; Lui, W.; Langworthy, M.; Tompkins, K.L.; Hatzopoulos, A.K.; Baldwin, H.S.; Zhou, B. Nfatc1 Coordinates Valve Endocardial Cell Lineage Development Required for Heart Valve Formation. Circ. Res. 2011, 109, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Shelton, E.L.; Yutzey, K.E. Tbx20 regulation of endocardial cushion cell proliferation and extracellular matrix gene expression. Dev. Biol. 2007, 302, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Zhang, W.; Hu, J.; Zhang, L.; Sultana, N.; Wu, B.; Cai, W.; Zhou, B.; Cai, C.-L. Tbx20 acts upstream of Wnt signaling to regulate endocardial cushion formation and valve remodeling during mouse cardiogenesis. Development 2013, 140, 3176–3187. [Google Scholar] [CrossRef] [Green Version]
- Boogerd, C.; Aneas, I.; Sakabe, N.; Dirschinger, R.J.; Cheng, Q.; Zhou, B.; Chen, J.; Nobrega, M.A.; Evans, S.M. Probing chromatin landscape reveals roles of endocardial TBX20 in septation. J. Clin. Investig. 2016, 126, 3023–3035. [Google Scholar] [CrossRef] [Green Version]
- Butcher, J.T.; McQuinn, T.C.; Sedmera, D.; Turner, D.; Markwald, R.R. Transitions in Early Embryonic Atrioventricular Valvular Function Correspond with Changes in Cushion Biomechanics That Are Predictable by Tissue Composition. Circ. Res. 2007, 100, 1503–1511. [Google Scholar] [CrossRef] [Green Version]
- Goddard, L.M.; Duchemin, A.-L.; Ramalingan, H.; Wu, B.; Chen, M.; Bamezai, S.; Yang, J.; Li, L.; Morley, M.P.; Wang, T.; et al. Hemodynamic Forces Sculpt Developing Heart Valves through a KLF2-WNT9B Paracrine Signaling Axis. Dev. Cell 2017, 43, 274–289.e5. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, A.; Yutzey, K.E. Mechanisms of heart valve development and disease. Development 2020, 147, dev183020. [Google Scholar] [CrossRef] [PubMed]
- Chiplunkar, A.R.; Lung, T.K.; Alhashem, Y.; Koppenhaver, B.A.; Salloum, F.; Kukreja, R.C.; Haar, J.L.; Lloyd, J.A. Krüppel-Like Factor 2 Is Required for Normal Mouse Cardiac Development. PLoS ONE 2013, 8, e54891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchemin, A.-L.; Vignes, H.; Vermot, J. Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. eLife 2019, 8, e44706. [Google Scholar] [CrossRef]
- Wünnemann, F.; MIBAVA Leducq Consortium Principal Investigators; Ta-Shma, A.; Preuss, C.; Leclerc, S.; Van Vliet, P.P.; Oneglia, A.; Thibeault, M.; Nordquist, E.; Lincoln, J.; et al. Loss of ADAMTS19 causes progressive non-syndromic heart valve disease. Nat. Genet. 2020, 52, 40–47. [Google Scholar] [CrossRef]
- Fukui, H.; Chow, R.W.-Y.; Xie, J.; Foo, Y.Y.; Yap, C.H.; Minc, N.; Mochizuki, N.; Vermot, J. Bioelectric signaling and the control of cardiac cell identity in response to mechanical forces. Science 2021, 374, 351–354. [Google Scholar] [CrossRef]
- Boselli, F.; Steed, E.; Freund, J.B.; Vermot, J. Anisotropic shear stress patterns predict the orientation of convergent tissue movements in the embryonic heart. Development 2017, 144, 4322–4327. [Google Scholar] [CrossRef] [Green Version]
- Vignes, H.; Vagena-Pantoula, C.; Prakash, M.; Fukui, H.; Norden, C.; Mochizuki, N.; Jug, F.; Vermot, J. Extracellular mechanical forces drive endocardial cell volume decrease during zebrafish cardiac valve morphogenesis. Dev. Cell 2022, 57, 598–609.e5. [Google Scholar] [CrossRef]
- Jeansson, M.; Gawlik, A.; Anderson, G.; Li, C.; Kerjaschki, D.; Henkelman, M.; Quaggin, S.E. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Investig. 2011, 121, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Dumont, D.J.; Gradwohl, G.; Fong, G.H.; Puri, M.C.; Gertsenstein, M.; Auerbach, A.; Breitman, M.L. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994, 8, 1897–1909. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Harmelink, C.; Baldwin, H.S. Tie2 regulates endocardial sprouting and myocardial trabeculation. JCI Insight 2019, 4, 96002. [Google Scholar] [CrossRef] [PubMed]
- Rentschler, S.; Vaidya, D.; Tamaddon, H.; Degenhardt, K.; Sassoon, D.; Morley, G.; Jalife, J.; Fishman, G. Visualization and functional characterization of the developing murine cardiac conduction system. Development 2001, 128, 1785–1792. [Google Scholar] [CrossRef]
- Rentschler, S.; Zander, J.; Meyers, K.; France, D.; Levine, R.; Porter, G.; Rivkees, S.A.; Morley, G.E.; Fishman, G.I. Neuregulin-1 promotes formation of the murine cardiac conduction system. Proc. Natl. Acad. Sci. USA 2002, 99, 10464–10469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shekhar, A.; Lin, X.; Liu, F.-Y.; Zhang, J.; Mo, H.; Bastarache, L.; Denny, J.; Cox, N.J.; Delmar, M.; Roden, D.M.; et al. Transcription factor ETV1 is essential for rapid conduction in the heart. J. Clin. Investig. 2016, 126, 4444–4459. [Google Scholar] [CrossRef] [Green Version]
- Bressan, M.; Yang, P.B.; Louie, J.D.; Navetta, A.M.; Garriock, R.J.; Mikawa, T. Reciprocal myocardial-endocardial interactions pattern the delay in atrioventricular junction conduction. Development 2014, 141, 4149–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, C.; Locke, A.E.; Feingold, E.; Reshey, B.; Espana, K.; Thusberg, J.; Mooney, S.; Bean, L.J.; Dooley, K.J.; Cua, C.L.; et al. An Excess of Deleterious Variants in VEGF-A Pathway Genes in Down-Syndrome-Associated Atrioventricular Septal Defects. Am. J. Hum. Genet. 2012, 91, 646–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, V.; Muth, A.N.; Ransom, J.F.; Schluterman, M.K.; Barnes, R.; King, I.N.; Grossfeld, P.D.; Srivastava, D. Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270–274. [Google Scholar] [CrossRef]
- Preuss, C.; Capredon, M.; Wünnemann, F.; Chetaille, P.; Prince, A.; Godard, B.; Leclerc, S.; Sobreira, N.; Ling, H.; Awadalla, P.; et al. Family Based Whole Exome Sequencing Reveals the Multifaceted Role of Notch Signaling in Congenital Heart Disease. PLoS Genet. 2016, 12, e1006335. [Google Scholar] [CrossRef]
- Laforest, B.; Andelfinger, G.; Nemer, M. Loss of Gata5 in mice leads to bicuspid aortic valve. J. Clin. Investig. 2011, 121, 2876–2887. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.C.; Zhao, Y.D.; Courtman, D.W.; Stewart, D.J. Abnormal Aortic Valve Development in Mice Lacking Endothelial Nitric Oxide Synthase. Circulation 2000, 101, 2345–2348. [Google Scholar] [CrossRef] [Green Version]
- Teekakirikul, P.; Zhu, W.; Gabriel, G.C.; Young, C.B.; Williams, K.; Martin, L.J.; Hill, J.C.; Richards, T.; Billaud, M.; Phillippi, J.A.; et al. Common deletion variants causing protocadherin-α deficiency contribute to the complex genetics of BAV and left-sided congenital heart disease. Hum. Genet. Genom. Adv. 2021, 2, 100037. [Google Scholar] [CrossRef] [PubMed]
- Sans-Coma, V.; Fernández, B.; Durán, A.C.; Thiene, G.; Arqué, J.M.; Muñoz-Chápuli, R.; Cardo, M. Fusion of Valve Cushions as a Key Factor in the Formation of Congenital Bicuspid Aortic Valves in Syrian Hamsters. Anat. Rec. 1996, 244, 490–498. [Google Scholar] [CrossRef]
- Henderson, D.J.; Eley, L.; Chaudhry, B. New Concepts in the Development and Malformation of the Arterial Valves. J. Cardiovasc. Dev. Dis. 2020, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Soto-Navarrete, M.T.; López-Unzu, M.A.; Durán, A.C.; Fernández, B. Embryonic development of bicuspid aortic valves. Prog. Cardiovasc. Dis. 2020, 63, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Fernández, B.; Soto-Navarrete, M.T.; López-García, A.; López-Unzu, M.A.; Durán, A.C.; Fernández, M.C. Bicuspid Aortic Valve in 2 Model Species and Review of the Literature. Vet. Pathol. 2020, 57, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.J.; Eley, L.; Turner, J.E.; Chaudhry, B. Development of the Human Arterial Valves: Understanding Bicuspid Aortic Valve. Front. Cardiovasc. Med. 2022, 8, 802930. [Google Scholar] [CrossRef] [PubMed]
- Durst, R.; Sauls, K.; Peal, D.S.; De Vlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef]
- Neri, T.; Hiriart, E.; van Vliet, P.; Faure, E.; Norris, R.A.; Farhat, B.; Jagla, B.; Lefrancois, J.; Sugi, Y.; Moore-Morris, T.; et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 2019, 10, 1929. [Google Scholar] [CrossRef]
- Liu, X.; Yagi, H.; Saeed, S.; Bais, A.S.; Gabriel, G.C.; Chen, Z.; Peterson, K.A.; Li, Y.; Schwartz, M.C.; Reynolds, W.T.; et al. The complex genetics of hypoplastic left heart syndrome. Nat. Genet. 2017, 49, 1152–1159. [Google Scholar] [CrossRef]
- Iascone, M.; Ciccone, R.; Galletti, L.; Marchetti, D.; Seddio, F.; Lincesso, A.R.; Pezzoli, L.; Vetro, A.; Barachetti, D.; Boni, L.; et al. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin. Genet. 2011, 81, 542–554. [Google Scholar] [CrossRef]
- Theis, J.L.; Hrstka, S.C.L.; Evans, J.M.; O’Byrne, M.M.; de Andrade, M.; O’Leary, P.W.; Nelson, T.J.; Olson, T.M. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Qual. Life Res. 2015, 134, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Liu, X.; Gabriel, G.C.; Wu, Y.; Peterson, K.; Murray, S.A.; Aronow, B.J.; Martin, L.J.; Benson, D.W.; Lo, C.W. The Genetic Landscape of Hypoplastic Left Heart Syndrome. Pediatr. Cardiol. 2018, 39, 1069–1081. [Google Scholar] [CrossRef]
- Miao, Y.; Tian, L.; Martin, M.; Paige, S.L.; Galdos, F.X.; Li, J.; Klein, A.; Zhang, H.; Ma, N.; Wei, Y.; et al. Intrinsic Endocardial Defects Contribute to Hypoplastic Left Heart Syndrome. Cell Stem Cell 2020, 27, 574–589.e8. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of rare inherited and de novo variants in 2871 congenital heart disease probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyndt, F.; Gueffet, J.-P.; Probst, V.; Jaafar, P.; Legendre, A.; Le Bouffant, F.; Toquet, C.; Roy, E.; McGregor, L.; Lynch, S.A.; et al. Mutations in the Gene Encoding Filamin A as a Cause for Familial Cardiac Valvular Dystrophy. Circulation 2007, 115, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, X.; Shen, A.; Jiao, W.; Guan, X.; Li, Z. GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur. J. Med. Genet. 2008, 51, 527–535. [Google Scholar] [CrossRef]
- Yang, Y.-Q.; Gharibeh, L.; Li, R.-G.; Xin, Y.-F.; Wang, J.; Liu, Z.-M.; Qiu, X.-B.; Xu, Y.-J.; Xu, L.; Qu, X.-K.; et al. GATA4 Loss-of-Function Mutations Underlie Familial Tetralogy of Fallot. Hum. Mutat. 2013, 34, 1662–1671. [Google Scholar] [CrossRef] [PubMed]
- Schott, J.-J.; Benson, D.W.; Basson, C.T.; Pease, W.; Silberbach, G.M.; Moak, J.P.; Maron, B.J.; Seidman, C.E.; Seidman, J.G. Congenital Heart Disease Caused by Mutations in the Transcription Factor NKX2–5. Science 1998, 281, 108–111. [Google Scholar] [CrossRef]
- Bauer, R.C.; Laney, A.O.; Smith, R.; Gerfen, J.; Morrissette, J.J.; Woyciechowski, S.; Garbarini, J.; Loomes, K.M.; Krantz, I.D.; Urban, Z.; et al. Jagged1 (JAG1) mutations in patients with tetralogy of fallot or pulmonic stenosis. Hum. Mutat. 2010, 31, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Baban, A.; Postma, A.; Marini, M.; Trocchio, G.; Santilli, A.; Pelegrini, M.; Sirleto, P.; Lerone, M.; Albanese, S.; Barnett, P.; et al. Identification of TBX5 mutations in a series of 94 patients with Tetralogy of Fallot. Am. J. Med. Genet. Part A 2014, 164, 3100–3107. [Google Scholar] [CrossRef]
- Klaassen, S.; Probst, S.; Oechslin, E.; Gerull, B.; Krings, G.; Schuler, P.; Greutmann, M.; Hürlimann, D.; Yegitbasi, M.; Pons, L.; et al. Mutations in Sarcomere Protein Genes in Left Ventricular Noncompaction. Circulation 2008, 117, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Hoedemaekers, Y.M.; Caliskan, K.; Michels, M.; Frohn-Mulder, I.; Van Der Smagt, J.J.; Phefferkorn, J.E.; Wessels, M.W.; Cate, F.T.; Sijbrands, E.; Dooijes, D.; et al. The Importance of Genetic Counseling, DNA Diagnostics, and Cardiologic Family Screening in Left Ventricular Noncompaction Cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, M.E.; Atkinson, D.L.; Ewart, A.K.; Morris, C.A.; Leppert, M.F.; Keating, M.T. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell 1993, 73, 159–168. [Google Scholar] [CrossRef]
- Guo, D.-C.; Papke, C.L.; Tran-Fadulu, V.; Regalado, E.S.; Avidan, N.; Johnson, R.J.; Kim, D.H.; Pannu, H.; Willing, M.C.; Sparks, E.; et al. Mutations in Smooth Muscle Alpha-Actin (ACTA2) Cause Coronary Artery Disease, Stroke, and Moyamoya Disease, Along with Thoracic Aortic Disease. Am. J. Hum. Genet. 2009, 84, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Yetman, A.T.; Starr, L.J.; Bleyl, S.B.; Meyers, L.; Delaney, J.W. Progressive Aortic Dilation Associated with ACTA2 Mutations Presenting in Infancy. Pediatrics 2015, 136, e262–e266. [Google Scholar] [CrossRef] [Green Version]
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.-P.; Lindsay, S.; D’Alessandro, L.C.A.; Swaminathan, G.J.; Bentham, J.; Arndt, A.-K.; et al. Rare Variants in NR2F2 Cause Congenital Heart Defects in Humans. Am. J. Hum. Genet. 2014, 94, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stittrich, A.-B.; Lehman, A.; Bodian, D.L.; Ashworth, J.; Zong, Z.; Li, H.; Lam, P.; Khromykh, A.; Iyer, R.K.; Vockley, J.G.; et al. Mutations in NOTCH1 Cause Adams-Oliver Syndrome. Am. J. Hum. Genet. 2014, 95, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Southgate, L.; Machado, R.D.; Snape, K.M.; Primeau, M.; Dafou, D.; Ruddy, D.M.; Branney, P.A.; Fisher, M.; Lee, G.J.; Simpson, M.A.; et al. Gain-of-Function Mutations of ARHGAP31, a Cdc42/Rac1 GTPase Regulator, Cause Syndromic Cutis Aplasia and Limb Anomalies. Am. J. Hum. Genet. 2011, 88, 574–585. [Google Scholar] [CrossRef] [Green Version]
- Blin, G.; Nury, D.; Stefanovic, S.; Neri, T.; Guillevic, O.; Brinon, B.; Bellamy, V.; Rücker-Martin, C.; Barbry, P.; Bel, A.; et al. A purified population of multipotent cardiovascular progenitors derived from primate pluripotent stem cells engrafts in postmyocardial infarcted nonhuman primates. J. Clin. Investig. 2010, 120, 1125–1139. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Xie, M.; Qiao, W.; Song, Y.; Zhang, Y.; Geng, Y.; Xu, W.; Wang, L.; Wang, Z.; Huang, K.; et al. Generation and characterization of cardiac valve endothelial-like cells from human pluripotent stem cells. Commun. Biol. 2021, 4, 1039. [Google Scholar] [CrossRef]
- Mikryukov, A.A.; Mazine, A.; Wei, B.; Yang, D.; Miao, Y.; Gu, M.; Keller, G.M. BMP10 Signaling Promotes the Development of Endocardial Cells from Human Pluripotent Stem Cell-Derived Cardiovascular Progenitors. Cell Stem Cell 2020, 28, 96–111.e7. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Israeli, Y.R.; Wasserman, A.H.; Gabalski, M.A.; Volmert, B.D.; Ming, Y.; Ball, K.A.; Yang, W.; Zou, J.; Ni, G.; Pajares, N.; et al. Self-assembling human heart organoids for the modeling of cardiac development and congenital heart disease. Nat. Commun. 2021, 12, 5142. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, P.; Jahnel, S.M.; Papai, N.; Giesshammer, M.; Deyett, A.; Schmidt, C.; Penc, M.; Tavernini, K.; Grdseloff, N.; Meledeth, C.; et al. Cardioids reveal self-organizing principles of human cardiogenesis. Cell 2021, 184, 3299–3317.e22. [Google Scholar] [CrossRef] [PubMed]
- Scherba, J.C.; Karra, R.; Turek, J.W.; Bursac, N. Toward improved understanding of cardiac development and congenital heart disease: The advent of cardiac organoids. J. Thorac. Cardiovasc. Surg. 2022. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cardiac Disease | Implicated Genes | Reference |
|---|---|---|
| Bicuspid aortic valve (BAV) | NOTCH1 GATA5 NOS3 PCDHA9 | Garg et al. [137] Laforest et al. [139] Lee et al. [140] Liu et al. [149], Yagi et al. [152] |
| Mitral valve prolapse (MVP) | FLNA DCHS1 | Kyndt et al. [155] Durst et al. [147] |
| Hypoplastic left heart syndrome (HLHS) | NOTCH1 PCDHA9 + SAP130 FN1 | Iascone et al. [150], Theis et al. [151] Liu et al. [149], Yagi et al. [152] Miao et al. [153] |
| Tetralogy of Fallot (TOF) | GATA4 NKX2-5 JAG1 TBX5 FLT4 | Zhang et al. [156] Yang et al. [157] Schott et al. [158] Bauer et al. [159] Baban et al. [160] Jin et al. [154] |
| Left ventricular non-compaction syndrome (LVNC) | MYH7, ACTC, TNNT2 MYBPC3, TNNI3, TPM1 | Klaassen et al. [161] Hoedemaekers et al. [162] |
| Aortic stenosis (AS) | ELN ADAMTS19 | Curran et al. [163] Wünnemann et al. [125] |
| Aortic dilation (AD) | ACTA2 | Guo et al. [164], Yetman et al. [165] |
| Complete AV canal defect (CAVC) | VEGFA NFR2F2 | Ackerman et al. [136] Al Turki et al. [166] |
| Adams-Oliver Syndrome (AOS) | NOTCH1 ARHGAP31 | Stittrich et al. [167] Southgate et al. [168] |
| Left ventricular outflow tract obstruction (LVOTO) | NOTCH1, ARHGAP31 | Preuss et al. [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feulner, L.; van Vliet, P.P.; Puceat, M.; Andelfinger, G. Endocardial Regulation of Cardiac Development. J. Cardiovasc. Dev. Dis. 2022, 9, 122. https://doi.org/10.3390/jcdd9050122
Feulner L, van Vliet PP, Puceat M, Andelfinger G. Endocardial Regulation of Cardiac Development. Journal of Cardiovascular Development and Disease. 2022; 9(5):122. https://doi.org/10.3390/jcdd9050122
Chicago/Turabian StyleFeulner, Lara, Patrick Piet van Vliet, Michel Puceat, and Gregor Andelfinger. 2022. "Endocardial Regulation of Cardiac Development" Journal of Cardiovascular Development and Disease 9, no. 5: 122. https://doi.org/10.3390/jcdd9050122